
Elias James Corey (born July 12, 1928) is an American organic chemist. In 1990, he won the Nobel Prize in Chemistry "for his development of the theory and methodology of organic synthesis", [3] specifically retrosynthetic analysis. [4] [5]
Elias James Corey (born July 12, 1928) is an American organic chemist. In 1990, he won the Nobel Prize in Chemistry "for his development of the theory and methodology of organic synthesis", [3] specifically retrosynthetic analysis. [4] [5]
E.J. Corey (the surname was anglicized from Levantine Arabic Khoury , meaning priest) was born to Lebanese Greek Orthodox Christian immigrants Fatima (née Hasham) and Elias Corey in Methuen, Massachusetts, 50 km (31 mi) north of Boston. [6] His mother changed his name from William to "Elias" to honor his father, who died eighteen months after Corey's birth. His widowed mother, brother, two sisters, aunt and uncle all lived together in a spacious house, struggling through the Great Depression. As a young boy, Corey was independent and enjoyed sports such as baseball, football, and hiking. He attended a Catholic elementary school and Lawrence High School in Lawrence, Massachusetts.
At the age of 16 Corey entered MIT, where he earned both a bachelor's degree in 1948 and a Ph.D. under Professor John C. Sheehan in 1951. Upon entering MIT, Corey's only experience with science was in mathematics, and he began his college career pursuing a degree in engineering. After his first chemistry class in his sophomore year he began rethinking his long-term career plans and graduated with a bachelor's degree in chemistry. Immediately thereafter, at the invitation of Professor John C. Sheehan, Corey remained at MIT for his Ph.D. After his graduate career he was offered an appointment at the University of Illinois at Urbana–Champaign, where he became a full professor of chemistry in 1956 at the age of 27. He was initiated as a member of the Zeta chapter of Alpha Chi Sigma at the University of Illinois in 1952. [7] In 1959, he moved to Harvard University, and in 1967 he was appointed the Sheldon Emory Professor and became a Guggenheim Fellow (1968-69). During his time at Harvard, Corey synthesised around 100 molecules, which previously had been only found in nature. [8] At Harvard, Corey is currently an emeritus professor of organic chemistry; his group maintains an active research program. He chose to work in organic chemistry because of "its intrinsic beauty and its great relevance to human health". [9] He has also been an advisor to Pfizer for more than 50 years. [10]
Among numerous honors, Corey was awarded the National Medal of Science in 1988, [11] the Nobel Prize in Chemistry in 1990, [5] and the American Chemical Society's greatest honor, the Priestley Medal, in 2004. [12]
Corey has developed several new synthetic reagents:

In the reaction, the alcohol nucleophilically displaces chlorine from the electropositive chromium(VI) metal. The chloride anion then acts as a base to afford the aldehyde product and chromium(IV).
The slightly acidic character of PCC makes it useful for cyclization reactions with alcohols and alkenes (Scheme 2). [14]

The initial oxidation yields the corresponding aldehyde, which can then undergo a Prins reaction with the neighboring alkene. After elimination and further oxidation, the product is a cyclic ketone. Conversely, powdered sodium acetate co-reagent inhibits reaction after formation of the aldehyde.
PCC's oxidative robustness has also rendered it useful in the realm of total synthesis (Scheme 3). This example illustrates the Babler oxidation reaction, in which tertiary allylic alcohols undergo a [3,3]-sigmatropic rearrangement. [15]
![[3,3] rearrangement with PCC PCC rearrangement3.png](http://upload.wikimedia.org/wikipedia/commons/thumb/8/87/PCC_rearrangement3.png/960px-PCC_rearrangement3.png)
Since 1972 the TBS group has become the most popular silicon protecting group (Scheme 4). [17] [18] TBS is stable to chromatography and labile enough to cleave under basic and acidic conditions. More importantly, TBS ethers are stable to such carbon nucleophiles as Grignard reagents and enolates. [19] [20] [21]
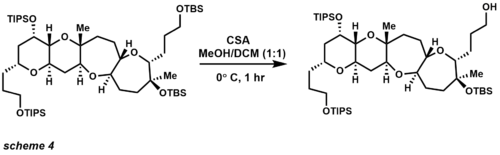
CSA (Camphorsulfonic acid) selectively removes a primary TBS ether in the presence of TIPS and tertiary TBS ethers. Other TBS deprotection methods include acids (also Lewis acids), and fluorides.
TIPS protecting groups provide increased selectivity of primary over secondary and tertiary alcohol protection. Their ethers are more stable under acidic and basic conditions than TBS ethers, but less labile for deprotection. [22] The most common cleavage reagents employ the same conditions as TBS ether, but longer reaction times.

Usually TBAF severs TBS ethers, but the hindered TBS ether above survives primary TIPS removal (scheme 5). [23]
The MEM protecting group was first described by Corey in 1976. [24] This protecting group is similar in reactivity and stability to other alkoxy methyl ethers under acidic conditions. Acidic conditions usually accomplish cleavage of MEM protecting groups, but coordination with metal halides greatly enhances lability (scheme 6). [25]

The formations of dithianes can be accomplished with a Lewis acid (scheme 7) or directly from carbonyl compounds. [27]

The pKa of dithianes is approximately 30, allowing deprotonation with an alkyl lithium reagent, typically n-butyllithium.
The reaction between dithianes and aldehydes is now known as the Corey-Seebach reaction. The dithiane, once deprotonated, serves as an acyl anion, attacking incoming electrophiles. Dithiane deprotection, usually with HgO, constructs a ketone product. [26]

Several reactions developed in Corey's lab have become commonplace in modern synthetic organic chemistry. At least 302 methods have been developed in the Corey group since 1950. [29] Several reactions have been named after him: Corey-Itsuno reduction, also known as the Corey-Bakshi-Shibata reduction, is an enantioselective reduction of ketones to alcohols through an oxazaborolidine catalyst, with various boranes as the stoichiometric reductant. [30]
The Corey group first demonstrated the catalyst's synthesis using borane and the chiral amino acid proline (scheme 9). [31] [32]

Later, Corey demonstrated that substituted boranes were easier to prepare and much more stable.
The reduction mechanism begins with the oxazaborolidine, only slightly basic at nitrogen, coordinating to a borane reductant (scheme 10). [32] Poor donation from the nitrogen to the boron leaves the Lewis acidity mostly intact, allowing coordination to the ketone substrate. The complexation of the substrate occurs from the most accessible lone pair of the oxygen, restricting rotation around the B-O bond due to the sterically neighboring phenyl group. [33]

Migration of the hydride from borane to the electrophilic ketone center occurs via a 6-membered ring transition state, leading to a four-membered ring intermediate, ultimately providing the chiral product and regeneration of the catalyst. [34]
The reaction has also been of great use to natural products chemists (scheme 11). [34] [35] The synthesis of dysidiolide by Corey and co-workers was achieved via an enantioselective CBS reduction using a borane-dimethylsulfide complex.

Corey-Fuchs alkyne synthesis is the synthesis of terminal alkynes through a one-carbon homologation of aldehydes using triphenylphosphine and carbon tetrabromide. [31] [36] The mechanism is similar to that of a combined Wittig reaction and Appel reaction. Reacting a phosphorus ylide formed in situ with the aldehyde substrate yields a dibromoolefin. [37]

On treatment with two equivalents of n-butyllithium, lithium halogen exchange and deprotonation yields a lithium acetylide species that undergoes hydrolysis to yield the terminal alkyne product (scheme 12). [31]
More recent developments include a modified procedure for one-pot synthesis. [38]
This synthetic transformation has been proven successful in the total synthesis (+)-taylorione by W.J. Kerr and co-workers (scheme 13). [39]

The Corey–Kim oxidation was a new conversion of alcohols into corresponding aldehydes and ketones. [31] [40] [41] This combination of N-chlorosuccinimidosulfonium chloride (NCS), dimethylsulfide (DMS), and triethylamine (TEA) offers a less toxic alternative to chromium-based oxidations.
The Corey-Kim reagent is formed in situ when the succinimide and sulfide react to form a dimethylsuccinimidosulfonium chloride species (scheme 14). [31]

Triethylamine deprotonates the alkoxysulfonium salt at the α position to afford the oxidized product. The reaction accommodates a wide array of functional groups, but allylic and benzylic alcohols are typically transformed into chlorides instead. [40]
Its application in synthesis is based on the mild protocol conditions and functional and protecting group compatibility. In the total synthesis of ingenol, Kuwajima and co-workers exploited the Corey-Kim oxidation by selectively oxidizing the less hindered secondary alcohol(scheme 15). [42]

Corey-Winter olefination is a stereospecific transformation of 1,2-diols to alkenes involving the diol substrate, thiocarbonyldiimidazole, and excess trialkylphosphite. [31] [43]
The exact mechanism is unknown, but has been narrowed down to two possible pathways. [44] The thionocarbonate and trialkylphosphite either form a phosphorus ylide or carbenoid intermediate.
The reaction is stereospecific for most substrates unless the product would lead to an exceedingly strained structure, as discovered when Corey et al attempted to form sterically hindered trans alkenes in certain 7-membered rings.
Stereospecfic alkenes are present in several natural products as the method continues to be exploited to yield a series of complex substrates. Professor T.K.M Shing et al used the Corey-Winter olefination reaction to synthesize (+)-Boesenoxide (scheme 16). [45]

CBS-type enantioselective Diels–Alder reaction has been developed using a similar scaffold to the enantioselective CBS reduction. [32] After the development of this reaction the CBS reagent proved to be a very versatile reagent for a series of several powerful synthetic transformations. The use of a chiral Lewis acid such as the CBS catalyst includes a broad range of unsaturated enones substrates.
The reaction likely proceeds via a highly organized 6-membered ring pre-transition state to deliver highly enantio-enriched products (scheme 17). [46]

This transition state likely occurs because of favorable pi-stacking with the phenyl substituent. [32] [47] The enantioselectivity of the process is facilitated from the diene approaching the dienophile from the opposite face of the phenyl substituent.
The Diels-Alder reaction is one of the most powerful transformations in synthetic chemistry. The synthesis of natural products using the Diels-Alder reaction as a transform has been applied especially to the formation of six-membered rings(scheme 18). [48]

Corey-Nicolaou macrolactonization provides the first method for preparing medium-to-large-size lactones. [31] [49] Previously, intermolecular outcompeted intramolecular lactonization even at low concentrations. One big advantage of this reaction is that it is performed under neutral conditions allowing the presence of acid and base-labile functional groups. As of 2016, rings of 7–44 members have been successfully synthesized using this method. [50] [51]

The reaction occurs in the presence of 2,2'-dipyridyl disulfide and triphenylphosphine with reflux of a nonpolar solvent such as benzene. The mechanism begins with formation of the 2-pyridinethiol ester (scheme 19). Proton-transfer provides a dipolar intermediate in which the alkoxide nucleophile attacks the electrophilic carbonyl center, providing a tetrahedral intermediate that yields the macrolactone product. [52]
One of the first examples of this protocol was applied to the total synthesis of zearalenone (scheme 20). [52]

The Johnson-Corey-Chaykovsky reaction synthesizes epoxides and cyclopropanes. [31] The reaction forms a sulfur ylide in situ that reacts with enones, ketones, aldehydes, and imines to form corresponding epoxides, cyclopropanes, and aziridines. [53] Two sulfur ylide variants have been employed that give different chemoselective products (scheme 21).The dimethylsulfoxonium methylide provides epoxides from ketones, but yields the cyclopropanes when enones are employed. Dimethylsulfonium methylide transforms ketones and enones to the corresponding epoxides. Dimethylsulfonium methylide is much more reactive and less stable than dimethylsulfoxonium methylide, so it is generated at low temperatures. [54]

Based on their reactivity, another distinct advantage of these two variants is that kinetically they provide a difference in diastereoselectivity. The reaction is very well established, and enantioselective variants (catalytic and stoichiometric) have also been achieved. From a retrosynthetic analysis standpoint, this reaction provides a reasonable alternative to conventional epoxidation reactions with alkenes (scheme 22). Danishefsky utilized this methodology for the synthesis of taxol. Diastereoselectivity is established by 1,3 interactions in the transition state required for epoxide closure. [55]

E. J. Corey and his research group have completed many total syntheses. At least 265 natural compounds have been synthesized in the Corey group since 1950. [56]
His 1969 total syntheses of several prostaglandins are considered classics. [57] [58] [59] [60] Specifically the synthesis of Prostaglandin F2α presents several challenges. The presence of both cis and trans olefins as well as five asymmetric carbon atoms renders the molecule a desirable challenge for organic chemists. Corey's retrosynthetic analysis outlines a few key disconnections that lead to simplified precursors (scheme 23).

Molecular simplification began first by disconnecting both carbon chains with a Wittig reaction and Horner-Wadsworth Emmons modification. The Wittig reaction affords the cis product, while the Horner-Wadsworth Emmons produces the trans olefin. The published synthesis reveals a 1:1 diastereomeric mixture of the carbonyl reduction using zinc borohydride. However, years later Corey and co-workers established the CBS reduction. One of the examples that exemplified this protocol was an intermediate in the prostaglandin synthesis revealing a 9:1 mixture of the desired diastereomer (scheme 24). [34]
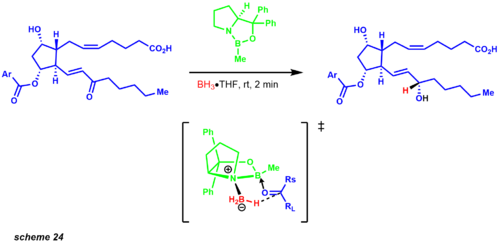
The iodolactonization transform affords an allylic alcohol leading to a key Baeyer-Villiger intermediate. This oxidation regioselectively inserts an oxygen atom between the ketone and the most electron-rich site. The pivotal intermediate leads to a straightforward conversion to the Diels-Alder structural goal, which provides the carbon framework for the functionalized cyclopentane ring. Later Corey developed an asymmetric Diels-Alder reaction employing a chiral oxazoborolidine, greatly simplifying the synthetic route to the prostaglandins.
Other notable syntheses:
Corey and his research group created LHASA, a program that uses artificial intelligence to discover sequences of reaction which may lead to total synthesis. [70] The program was one of the first to use a graphical interface to input and display chemical structures. [71]
E.J. Corey has more than 1100 publications. [72] In 2002, the American Chemical Society (ACS) recognized him as the "Most Cited Author in Chemistry". In 2007, he received the first ACS Publications Division "Cycle of Excellence High Impact Contributor Award" [73] and was ranked the number one chemist in terms of research impact by the Hirsch Index (h-index). [74] His books include:
Jason Altom, one of Corey's students, committed suicide in 1998. [75] Altom's suicide was well-known because of his suicide notes, which called for protection of students from "abusive research advisors," which he stated were a part of the reason he killed himself. One of the letters was addressed to Corey. Altom's suicide note also contained explicit instructions on how to reform the relationship between students and their supervisors. [76]
Corey was reportedly devastated and bewildered by his student's death. [77] Corey said, "That letter doesn't make sense. At the end, Jason must have been delusional or irrational in the extreme." Corey also claimed he never questioned Altom's intellectual contributions. "I did my best to guide Jason as a mountain guide would to guide someone climbing a mountain. I did my best every step of the way," Corey states. "My conscience is clear. Everything Jason did came out of our partnership. We never had the slightest disagreement." [75] Students and professors said Altom actually retained Corey's support even as Altom struggled to make progress. [77]
As of 2010, approximately 700 people have been Corey Group members including notable students Eric Block, Dale L. Boger, Weston T. Borden, David E. Cane, Rick L. Danheiser, William L. Jorgensen, John Katzenellenbogen, Alan P. Kozikowski, Bruce H. Lipshutz, David R. Liu, Albert Meyers, K. C. Nicolaou, Ryōji Noyori, Gary H. Posner, Bengt I. Samuelsson, Dieter Seebach, Vinod K. Singh, Brian Stoltz, Alice Ting, Hisashi Yamamoto, Phil Baran and Jin-Quan Yu. A database of 580 former members and their current affiliation was developed for Corey's 80th birthday in July 2008. [78]
When awarded the Priestley Medal in 2004, E. J. Corey created a controversy with his claim to have inspired Robert Burns Woodward prior to the development of the Woodward–Hoffmann rules. Corey wrote:
"On May 4, 1964, I suggested to my colleague R. B. Woodward a simple explanation involving the symmetry of the perturbed (HOMO) molecular orbitals for the stereoselective cyclobutene → 1,3-butadiene and 1,3,5-hexatriene → cyclohexadiene conversions that provided the basis for the further development of these ideas into what became known as the Woodward–Hoffmann rules." [79]
This was Corey's first public statement on his claim that starting on May 5, 1964, Woodward put forth Corey's explanation as his own thought with no mention of Corey and the conversation of May 4. Corey had discussed his claim privately with Hoffmann and close colleagues since 1964. Corey mentions that he made the Priestley statement "so the historical record would be correct". [80]
Corey's claim and contribution were publicly rebutted by Roald Hoffmann in the journal Angewandte Chemie . In the rebuttal, Hoffmann states that he asked Corey over the course of their long discussion of the matter why Corey did not make the issue public. Corey responded that he thought such a public disagreement would hurt Harvard and that he would not "consider doing anything against Harvard, to which I was and am so devoted." Corey also hoped that Woodward himself would correct the historical record "as he grew older, more considerate, and more sensitive to his own conscience." [81] Woodward died suddenly of a heart attack in his sleep in 1979.
E.J. Corey has received more than 40 major awards including the Linus Pauling Award (1973), Franklin Medal (1978), Tetrahedron Prize (1983), Wolf Prize in Chemistry (1986), National Medal of Science (1988), Japan Prize (1989), Nobel Prize in Chemistry (1990), Golden Plate Award of the American Academy of Achievement (1991), [82] Roger Adams Award (1993), and the Priestley Medal (2004). [12] He was inducted into the Alpha Chi Sigma Hall of Fame in 1998. [7] As of 2008, he has been awarded 19 honorary degrees from universities around the world including Oxford University (UK), Cambridge University (UK), and National Chung Cheng University. [83] In 2013, the E.J. Corey Institute of Biomedical Research (CIBR) opened in Jiangyin, Jiangsu Province, China. [84]
Corey was elected a Foreign Member of the Royal Society (ForMemRS) in 1998. [2]
{{cite web}}: CS1 maint: bot: original URL status unknown (link), The Boston Globe via Archive.org (January 2, 2001).| International | |
|---|---|
| National | |
| Academics | |
| People | |
| Other | |
