
Bortezomib, sold under the brand name Velcade among others, is an anti-cancer medication used to treat multiple myeloma and mantle cell lymphoma. This includes multiple myeloma in those who have and have not previously received treatment. It is generally used together with other medications. It is given by injection.
Proteasome inhibitors are drugs that block the action of proteasomes, cellular complexes that break down proteins. They are being studied in the treatment of cancer; and three are approved for use in treating multiple myeloma.
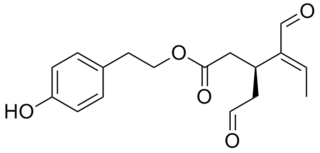
Oleocanthal is a phenylethanoid, or a type of natural phenolic compound found in extra-virgin olive oil. It appears to be responsible for the burning sensation that occurs in the back of the throat when consuming such oil. Oleocanthal is a tyrosol ester and its chemical structure is related to oleuropein, also found in olive oil.
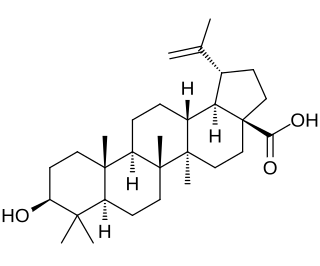
Betulinic acid is a naturally occurring pentacyclic triterpenoid which has antiretroviral, antimalarial, and anti-inflammatory properties, as well as a more recently discovered potential as an anticancer agent, by inhibition of topoisomerase. It is found in the bark of several species of plants, principally the white birch from which it gets its name, but also the ber tree, selfheal, the tropical carnivorous plants Triphyophyllum peltatum and Ancistrocladus heyneanus, Diospyros leucomelas, a member of the persimmon family, Tetracera boiviniana, the jambul, flowering quince, rosemary, and Pulsatilla chinensis.
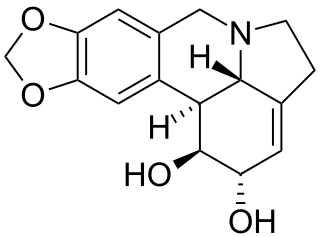
Lycorine is a toxic crystalline alkaloid found in various Amaryllidaceae species, such as the cultivated bush lily, surprise lilies (Lycoris), and daffodils (Narcissus). It may be highly poisonous, or even lethal, when ingested in certain quantities. Regardless, it is sometimes used medicinally, a reason why some groups may harvest the very popular Clivia miniata.

Nereus Pharmaceuticals was a pharmaceutical company focused on the development of natural products from marine microbial and other natural sources into small molecule human therapeutics. The major disease area addressed by Nereus is cancer. Nereus was purchased by Triphase Research and Development in 2012.

Indolocarbazoles (ICZs) are a class of compounds that are under current study due to their potential as anti-cancer drugs and the prospective number of derivatives and uses found from the basic backbone alone. First isolated in 1977, a wide range of structures and derivatives have been found or developed throughout the world. Due to the extensive number of structures available, this review will focus on the more important groups here while covering their occurrence, biological activity, biosynthesis, and laboratory synthesis.

Kendomycin is an anticancer macrolide first isolated from Streptomyces violaceoruber. It has potent activity as an endothelin receptor antagonist and anti-osteoporosis agent. It also has strong cytotoxicity against various tumor cell lines.

Carfilzomib, sold under the brand name Kyprolis, is an anti-cancer medication acting as a selective proteasome inhibitor. Chemically, it is a tetrapeptide epoxyketone and an analog of epoxomicin. It was developed by Onyx Pharmaceuticals.

Callystatin A is a polyketide natural product from the leptomycin family of secondary metabolites. It was first isolated in 1997 from the marine sponge Callyspongia truncata which was collected from the Goto Islands in the Nagasaki Prefecture of Japan by the Kobayashi group. Since then its absolute configuration has been elucidated and callystatin A was discovered to have anti-fungal and anti-tumor activities with extreme potency against the human epidermoid carcinoma KB cells (IG50 = 10 pg/ml) and the mouse lymphocytic leukemia Ll210 cells (IG50 = 20 pg/ml).
The salinosporamides are a group of closely related chemical compounds isolated from marine bacteria in the genus Salinispora. They possess a densely functionalized γ-lactam-β-lactone bicyclic core.
Cylindrocyclophanes are a class of cyclophane, a group of aromatic hydrocarbons composed of two benzene rings attached in a unique structure. Cylindrocyclophanes were the first cyclophanes found in nature, isolated from a species of cyanobacteria, and have proven to be an interesting group of compounds to study due to their unusual molecular structure and intriguing biological possibilities, especially its cytotoxicity to some cancer cell lines.

Fellutamides are tripeptide derivatives from Penicillium fellutanum and other fungi. They are potent proteasome inhibitor that stimulates nerve growth factor synthesis in vitro.
Salinispora tropica is an obligate marine actinomycete bacterium species. It produces salinosporamide A and salinosporamide B, potential anti-cancer agents, as well as the polycyclic macrolides sporolide A and B.

Eudistomins are β-carboline derivatives, isolated from ascidians, like Ritterella sigillinoides, Lissoclinum fragile, or Pseudodistoma aureum.

Salinispora is a genus of obligately aerobic, gram-positive, non-acid-fast bacteria belonging to the family of Micromonosporaceae. They are heterotrophic, non-motile, and obligately grow under high osmotic/ionic-strength conditions. They are the first identified genus of gram-positive bacteria which has a high osmotic/ionic-strength requirement for survival. They are widely abundant in tropical marine sediments and were first identified in 2002. This genus of bacteria has potential biotechnological significance due to their production of novel secondary metabolites which can be used pharmaceutically.

Dideoxyverticillin A, also known as (+)-11,11′-dideoxyverticillin A, is a complex epipolythiodioxopiperazine initially isolated from the marine fungus Penicillium sp. in 1999. It has also been found in the marine fungus Bionectriaceae, and belongs to a class of naturally occurring 2,5-diketopiperazines.
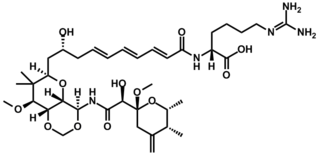
Onnamide A is a bioactive natural product found in Theonella swinhoei, a species of marine sponge whose genus is well known for yielding a diverse set of biologically active natural products, including the swinholides and polytheonamides. It bears structural similarities to the pederins, a family of compounds known to inhibit protein synthesis in eukaryotic cells. Onnamide A and its analogues have attracted academic interest due to their cytotoxicity and potential for combating the growth and proliferation of cancer cells.
Steroidal aromatase inhibitors are a class of drugs that are mostly used for treating breast cancer in postmenopausal women. High levels of estrogen in breast tissue increases the risk of developing breast cancer and the enzyme aromatase is considered to be a good therapeutic target when treating breast cancer due to it being involved in the final step of estrogen biosynthetic pathway and also its inhibition will not affect production of other steroids. Aromatase Inhibitors are classified into two categories based on their structure, nonsteroidal and steroidal; the latter resemble the structure of androstenedione. Steroidal aromatase inhibitors irreversibly inhibit the enzyme by binding covalently to the binding site of aromatase so the substrate cannot access it.

Oprozomib is an orally active second-generation proteasome inhibitor developed by Proteolix, which was acquired by Onyx Pharmaceuticals, an Amgen subsidiary, in 2009. It selectively inhibits chymotrypsin-like activity of both the constitutive proteasome (PSMB5) and immunoproteasome (LMP7).



