A chemical database is a database specifically designed to store chemical information. This information is about chemical and crystal structures, spectra, reactions and syntheses, and thermophysical data.
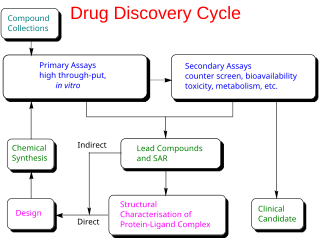
In the fields of medicine, biotechnology and pharmacology, drug discovery is the process by which new candidate medications are discovered.
Cheminformatics refers to use of physical chemistry theory with computer and information science techniques—so called "in silico" techniques—in application to a range of descriptive and prescriptive problems in the field of chemistry, including in its applications to biology and related molecular fields. Such in silico techniques are used, for example, by pharmaceutical companies and in academic settings to aid and inform the process of drug discovery, for instance in the design of well-defined combinatorial libraries of synthetic compounds, or to assist in structure-based drug design. The methods can also be used in chemical and allied industries, and such fields as environmental science and pharmacology, where chemical processes are involved or studied.

UniProt is a freely accessible database of protein sequence and functional information, many entries being derived from genome sequencing projects. It contains a large amount of information about the biological function of proteins derived from the research literature. It is maintained by the UniProt consortium, which consists of several European bioinformatics organisations and a foundation from Washington, DC, United States.
The European Bioinformatics Institute (EMBL-EBI) is an Intergovernmental Organization (IGO) which, as part of the European Molecular Biology Laboratory (EMBL) family, focuses on research and services in bioinformatics. It is located on the Wellcome Genome Campus in Hinxton near Cambridge, and employs over 600 full-time equivalent (FTE) staff. Institute leaders such as Rolf Apweiler, Alex Bateman, Ewan Birney, and Guy Cochrane, an adviser on the National Genomics Data Center Scientific Advisory Board, serve as part of the international research network of the BIG Data Center at the Beijing Institute of Genomics.
PubChem is a database of chemical molecules and their activities against biological assays. The system is maintained by the National Center for Biotechnology Information (NCBI), a component of the National Library of Medicine, which is part of the United States National Institutes of Health (NIH). PubChem can be accessed for free through a web user interface. Millions of compound structures and descriptive datasets can be freely downloaded via FTP. PubChem contains multiple substance descriptions and small molecules with fewer than 100 atoms and 1,000 bonds. More than 80 database vendors contribute to the growing PubChem database.
A structural analog, also known as a chemical analog or simply an analog, is a compound having a structure similar to that of another compound, but differing from it in respect to a certain component.
Chemical Entities of Biological Interest, also known as ChEBI, is a chemical database and ontology of molecular entities focused on 'small' chemical compounds, that is part of the Open Biomedical Ontologies (OBO) effort at the European Bioinformatics Institute (EBI). The term "molecular entity" refers to any "constitutionally or isotopically distinct atom, molecule, ion, ion pair, radical, radical ion, complex, conformer, etc., identifiable as a separately distinguishable entity". The molecular entities in question are either products of nature or synthetic products which have potential bioactivity. Molecules directly encoded by the genome, such as nucleic acids, proteins and peptides derived from proteins by proteolytic cleavage, are not as a rule included in ChEBI.

Chemogenomics, or chemical genomics, is the systematic screening of targeted chemical libraries of small molecules against individual drug target families with the ultimate goal of identification of novel drugs and drug targets. Typically some members of a target library have been well characterized where both the function has been determined and compounds that modulate the function of those targets have been identified. Other members of the target family may have unknown function with no known ligands and hence are classified as orphan receptors. By identifying screening hits that modulate the activity of the less well characterized members of the target family, the function of these novel targets can be elucidated. Furthermore, the hits for these targets can be used as a starting point for drug discovery. The completion of the human genome project has provided an abundance of potential targets for therapeutic intervention. Chemogenomics strives to study the intersection of all possible drugs on all of these potential targets.
The Structural Genomics Consortium (SGC) is a public-private-partnership focusing on elucidating the functions and disease relevance of all proteins encoded by the human genome, with an emphasis on those that are relatively understudied. The SGC places all its research output into the public domain without restriction and does not file for patents and continues to promote open science. Two recent publications revisit the case for open science. Founded in 2003, and modelled after the Single Nucleotide Polymorphism Database (dbSNP) Consortium, the SGC is a charitable company whose Members comprise organizations that contribute over $5,4M Euros to the SGC over a five-year period. The Board has one representative from each Member and an independent Chair, who serves one 5-year term. The current Chair is Anke Müller-Fahrnow (Germany), and previous Chairs have been Michael Morgan (U.K.), Wayne Hendrickson (U.S.A.), Markus Gruetter (Switzerland) and Tetsuyuki Maruyama (Japan). The founding and current CEO is Aled Edwards (Canada). The founding Members of the SGC Company were the Canadian Institutes of Health Research, Genome Canada, the Ontario Research Fund, GlaxoSmithKline and Wellcome Trust. The current Members comprise Bayer Pharma AG, Bristol Myers Squibb, Boehringer Ingelheim, the Eshelman Institute for Innovation, Genentech, Genome Canada, Janssen, Merck KGaA, Pfizer, and Takeda.
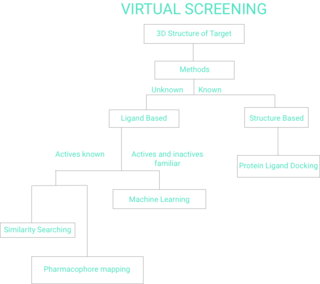
Virtual screening (VS) is a computational technique used in drug discovery to search libraries of small molecules in order to identify those structures which are most likely to bind to a drug target, typically a protein receptor or enzyme.

ChemSpider is a database of chemicals. ChemSpider is owned by the Royal Society of Chemistry.

Collaborative Drug Discovery (CDD) is a software company founded in 2004 as a spin-out of Eli Lilly by Barry Bunin, PhD. CDD utilizes a web-based database solution for managing drug discovery data, primarily through the CDD Vault product which is focused around small molecules and associated bio-assay data. In 2021, CDD launched its first commercial data offering, PharmaKB, formerly BioHarmony, as The Pharma KnowledgeBase, which is centered around pharma company, drug, and disease information for research, business intelligence, and investors.
Druggability is a term used in drug discovery to describe a biological target that is known to or is predicted to bind with high affinity to a drug. Furthermore, by definition, the binding of the drug to a druggable target must alter the function of the target with a therapeutic benefit to the patient. The concept of druggability is most often restricted to small molecules but also has been extended to include biologic medical products such as therapeutic monoclonal antibodies.

The TDR Targets database is a bioinformatics project that seeks to exploit the availability of diverse genomic and chemical datasets to facilitate the identification and prioritization of drugs and drug targets in neglected disease pathogens. TDR in the name of the database stands from the popular abbreviation for a special programme within the World Health Organization, whose focus is Tropical Disease Research. The project was jumpstarted by funds from this programme, and the initial focus of the resource was on organisms/diseases of high priority for this Programme.
The IUPHAR/BPS Guide to PHARMACOLOGY is an open-access website, acting as a portal to information on the biological targets of licensed drugs and other small molecules. The Guide to PHARMACOLOGY is developed as a joint venture between the International Union of Basic and Clinical Pharmacology (IUPHAR) and the British Pharmacological Society (BPS). This replaces and expands upon the original 2009 IUPHAR Database. The Guide to PHARMACOLOGY aims to provide a concise overview of all pharmacological targets, accessible to all members of the scientific and clinical communities and the interested public, with links to details on a selected set of targets. The information featured includes pharmacological data, target, and gene nomenclature, as well as curated chemical information for ligands. Overviews and commentaries on each target family are included, with links to key references.

Experimental factor ontology, also known as EFO, is an open-access ontology of experimental variables particularly those used in molecular biology. The ontology covers variables which include aspects of disease, anatomy, cell type, cell lines, chemical compounds and assay information. EFO is developed and maintained at the EMBL-EBI as a cross-cutting resource for the purposes of curation, querying and data integration in resources such as Ensembl, ChEMBL and Expression Atlas.

Professor Bissan Al-Lazikani PhD FRSB MBCS is a data scientist and drug discoverer. She applies computational techniques to help solve critical bottlenecks in cancer drug discovery and development. Since 2021 she has been professor of genomic medicine at The University of Texas MD Anderson Cancer Center.
Target 2035 is a global effort or movement to discover open science, pharmacological modulator(s) for every protein in the human proteome by the year 2035. The effort is led by the Structural Genomics Consortium with the intention that this movement evolves organically. Target 2035 has been borne out of the success that chemical probes have had in elevating or de-prioritizing the therapeutic potential of protein targets. The availability of open access pharmacological tools is a largely unmet aspect of drug discovery especially for the dark proteome.
