Elias James Corey is an American organic chemist. In 1990, he won the Nobel Prize in Chemistry "for his development of the theory and methodology of organic synthesis", specifically retrosynthetic analysis. Regarded by many as one of the greatest living chemists, he has developed numerous synthetic reagents, methodologies and total syntheses and has advanced the science of organic synthesis considerably.
An ylide or ylid is a neutral dipolar molecule containing a formally negatively charged atom (usually a carbanion) directly attached to a heteroatom with a formal positive charge (usually nitrogen, phosphorus or sulfur), and in which both atoms have full octets of electrons. The result can be viewed as a structure in which two adjacent atoms are connected by both a covalent and an ionic bond; normally written X+–Y−. Ylides are thus 1,2-dipolar compounds, and a subclass of zwitterions. They appear in organic chemistry as reagents or reactive intermediates.
In organic chemistry, a nucleophilic addition reaction is an addition reaction where a chemical compound with an electrophilic double or triple bond reacts with a nucleophile, such that the double or triple bond is broken. Nucleophilic additions differ from electrophilic additions in that the former reactions involve the group to which atoms are added accepting electron pairs, whereas the latter reactions involve the group donating electron pairs.
In organic chemistry, hydrocyanation is a process for conversion of alkenes to nitriles. The reaction involves the addition of hydrogen cyanide and requires a catalyst. This conversion is conducted on an industrial scale for the production of precursors to nylon.
The Wittig reaction or Wittig olefination is a chemical reaction of an aldehyde or ketone with a triphenyl phosphonium ylide called a Wittig reagent. Wittig reactions are most commonly used to convert aldehydes and ketones to alkenes. Most often, the Wittig reaction is used to introduce a methylene group using methylenetriphenylphosphorane (Ph3P=CH2). Using this reagent, even a sterically hindered ketone such as camphor can be converted to its methylene derivative.

Nucleophilic conjugate addition is a type of organic reaction. Ordinary nucleophilic additions or 1,2-nucleophilic additions deal mostly with additions to carbonyl compounds. Simple alkene compounds do not show 1,2 reactivity due to lack of polarity, unless the alkene is activated with special substituents. With α,β-unsaturated carbonyl compounds such as cyclohexenone it can be deduced from resonance structures that the β position is an electrophilic site which can react with a nucleophile. The negative charge in these structures is stored as an alkoxide anion. Such a nucleophilic addition is called a nucleophilic conjugate addition or 1,4-nucleophilic addition. The most important active alkenes are the aforementioned conjugated carbonyls and acrylonitriles.
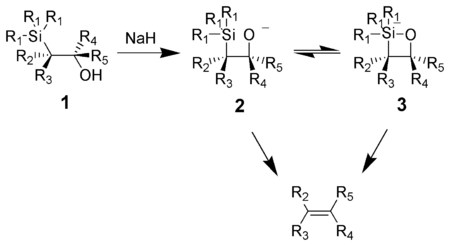
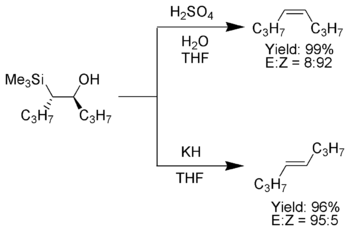
In organic chemistry the Brook rearrangement refers to any [1,n] carbon to oxygen silyl migration. The rearrangement was first observed in the late 1950s by Canadian chemist Adrian Gibbs Brook (1924–2013), after which the reaction is named. These migrations can be promoted in a number of different ways, including thermally, photolytically or under basic/acidic conditions. In the forward direction, these silyl migrations produce silyl ethers as products which is driven by the stability of the oxygen-silicon bond.
The Horner–Wadsworth–Emmons (HWE) reaction is a chemical reaction used in organic chemistry of stabilized phosphonate carbanions with aldehydes to produce predominantly E-alkenes.

Tebbe's reagent is the organometallic compound with the formula (C5H5)2TiCH2ClAl(CH3)2. It is used in the methylidenation of carbonyl compounds, that is it converts organic compounds containing the R2C=O group into the related R2C=CH2 derivative. It is a red solid that is pyrophoric in the air, and thus is typically handled with air-free techniques. It was originally synthesized by Fred Tebbe at DuPont Central Research.

The Darzens reaction is the chemical reaction of a ketone or aldehyde with an α-haloester in the presence of a base to form an α,β-epoxy ester, also called a "glycidic ester". This reaction was discovered by the organic chemist Auguste Georges Darzens in 1904.
The Julia olefination (also known as the Julia–Lythgoe olefination) is the chemical reaction used in organic chemistry of phenyl sulfones (1) with aldehydes (or ketones) to give alkenes (olefins)(3) after alcohol functionalization and reductive elimination using sodium amalgam or SmI2. The reaction is named after the French chemist Marc Julia.

Organozinc chemistry is the study of the physical properties, synthesis, and reactions of organozinc compounds, which are organometallic compounds that contain carbon (C) to zinc (Zn) chemical bonds.
The Rubottom oxidation is a useful, high-yielding chemical reaction between silyl enol ethers and peroxyacids to give the corresponding α-hydroxy carbonyl product. The mechanism of the reaction was proposed in its original disclosure by A.G. Brook with further evidence later supplied by George M. Rubottom. After a Prilezhaev-type oxidation of the silyl enol ether with the peroxyacid to form the siloxy oxirane intermediate, acid-catalyzed ring-opening yields an oxocarbenium ion. This intermediate then participates in a 1,4-silyl migration to give an α-siloxy carbonyl derivative that can be readily converted to the α-hydroxy carbonyl compound in the presence of acid, base, or a fluoride source.
Selenoxide elimination is a method for the chemical synthesis of alkenes from selenoxides. It is most commonly used to synthesize α,β-unsaturated carbonyl compounds from the corresponding saturated analogues. It is mechanistically related to the Cope reaction.
The Abramov reaction is the related conversions of trialkyl to α-hydroxy phosphonates by the addition to carbonyl compounds. In terms of mechanism, the reaction involves attack of the nucleophilic phosphorus atom on the carbonyl carbon. It was named after the Russian chemist Vasilii Semenovich Abramov (1904–1968) in 1957.
The [2,3]-Wittig rearrangement is the transformation of an allylic ether into a homoallylic alcohol via a concerted, pericyclic process. Because the reaction is concerted, it exhibits a high degree of stereocontrol, and can be employed early in a synthetic route to establish stereochemistry. The Wittig rearrangement requires strongly basic conditions, however, as a carbanion intermediate is essential. [1,2]-Wittig rearrangement is a competitive process.

Reductions with samarium(II) iodide involve the conversion of various classes of organic compounds into reduced products through the action of samarium(II) iodide, a mild one-electron reducing agent.
Desulfonylation reactions are chemical reactions leading to the removal of a sulfonyl group from organic compounds. As the sulfonyl functional group is electron-withdrawing, methods for cleaving the sulfur–carbon bonds of sulfones are typically reductive in nature. Olefination or replacement with hydrogen may be accomplished using reductive desulfonylation methods.

Manganese-mediated coupling reactions are radical coupling reactions between enolizable carbonyl compounds and unsaturated compounds initiated by a manganese(III) salt, typically manganese(III) acetate. Copper(II) acetate is sometimes used as a co-oxidant to assist in the oxidation of intermediate radicals to carbocations.
William Clark Still is an American organic chemist. As a distinguished professor at Columbia University, Clark Still made significant contributions to the field of organic chemistry, particularly in the areas of natural product synthesis, reaction development, conformational analysis, macrocyclic stereocontrol, and computational chemistry. Still and coworkers also developed the purification technique known as flash column chromatography which is widely used for the purification of organic compounds.