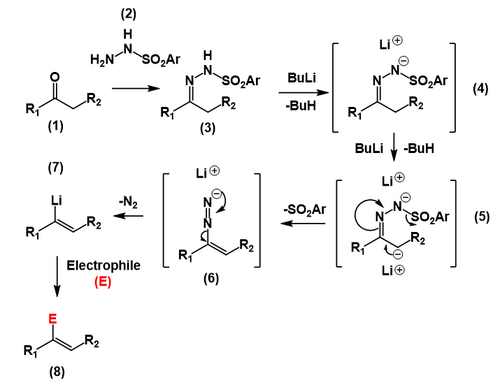
In a prelude to the actual Shapiro reaction, a ketone or an aldehyde is converted to the tosylhydrazone. Two equivalents of n-butyllithium gives the carbanion, which produces a carbon–carbon double bond, ejecting the tosyl anion. The resulting diazonium anion loses molecular nitrogen giving the vinyllithium species.
The reaction's directionality is controlled by the stereochemistry of the hydrazone, with deprotonation occurring cis to the tosylamide group. This is due to coordination by the nitrogen atom.[6]
Intermediate explaining directionality of the Shapiro reaction
Scope
The position of the alkene in the product is controlled by the site of deprotonation by the organolithium base. In general, the kinetically favored, less substituted site of differentially substituted tosylhydrazones is deprotonated selectively, leading to the less substituted vinyllithium intermediate. Although many secondary reactions exist for the vinyllithium functional group, in the Shapiro reaction in particular water is added, resulting in protonation to the alkene.[7] Other reactions of vinyllithium compounds include alkylation reactions with for instance alkyl halides.[8]
Shapiro reactions starting from camphor (1) through the intermediate hydrazone (2) to the vinyllithium (3). Addition of water (c) results in 2-bornene (4) and addition of an alkyl bromide (d) gives 5
Importantly, the Shapiro reaction cannot be used to synthesize 1-lithioalkenes (and the resulting functionalized derivatives), as sulfonylhydrazones derived from aldehydes undergo exclusive addition of the organolithium base to the carbon of the C–N double bond.[9]
Catalytic Shapiro reaction
Traditional Shapiro reactions require stoichiometric (sometimes excess) amounts of base to generate the alkenyllithium reagents. To combat this problem, Yamamoto and coworkers developed an efficient stereoselective and regioselective route to alkenes using a combination of ketone phenylaziridinylhydrazones as arenesulfonylhydrazone equivalents with a catalytic amount of lithium amides. The required phenylaziridinylhydrazone was prepared from the condensation of undecan-6-one with 1-amino-2-phenylaziridine. Treatment of the phenylaziridinylhydrazone with 0.3 equivalents of LDA in ether resulted in the alkene shown below with a cis:trans ratio of 99.4:0.6. The ratio was determined by capillary GLC analysis after conversion to the corresponding epoxides with mCPBA. The catalyst loading can be reduced to 0.05 equivalents in the case of a 30mmol scale reaction.
The high stereoselectivity is obtained by the preferential abstraction of the α-methylene hydrogen syn to the phenylaziridine, and is also accounted for by the internal chelation of the lithiated intermediated.[10]
The Shapiro reaction with N-aziridinylhydrazones produces the alkene product, as well as stryrene and gaseous nitrogen as byproducts. The cycle of the catalytic Shapiro reaction is also shown.
A one pot in situ combined Shapiro-Suzuki reaction
The Shapiro reaction can also be combined with the Suzuki reaction to produce a variety of olefin products. Keay and coworkers have developed methodology that combines these reactions in a one pot process that does not require the isolation of the boronic acid, a setback of the traditional Suzuki coupling. This reaction has a wide scope, tolerating a slew of trisylhydrazones and aryl halides, as well as several solvents and Pd sources.[11]
The Shapiro and Suzuki reactions are combined to yield a variety of alkene products.
An application of the Shapiro reaction in total synthesis
The Shapiro reaction has been used to generate olefins towards to complex natural products. K. Mori and coworkers wanted to determine the absolute configuration of the phytocassane group of a class of natural products called phytoalexins. This was accomplished by preparing the naturally occurring (–)-phytocassane D from (R)-Wieland-Miescher ketone. On the way to (–)-phytocassane D, a tricyclic ketone was subjected to Shapiro reaction conditions to yield the cyclic alkene product. [12]
Use of the Shapiro reaction in the synthesis of (–)-phytocassane D
↑ Shapiro, R. H.; Lipton, M. F.; Kolonko, K. J.; Buswell, R. L.; Capuano, L. A. (1975). "Tosylhydrazones and alkyllithium reagents: More on the regiospecificity of the reaction and the trapping of three intermediates". Tetrahedron Lett.16 (22–23): 1811–1814. doi:10.1016/S0040-4039(00)75263-4.
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.