In organic chemistry, an alkene, or olefin, is a hydrocarbon containing a carbon–carbon double bond. The double bond may be internal or in the terminal position. Terminal alkenes are also known as α-olefins.
Hydroboration–oxidation reaction is a two-step hydration reaction that converts an alkene into an alcohol. The process results in the syn addition of a hydrogen and a hydroxyl group where the double bond had been. Hydroboration–oxidation is an anti-Markovnikov reaction, with the hydroxyl group attaching to the less-substituted carbon. The reaction thus provides a more stereospecific and complementary regiochemical alternative to other hydration reactions such as acid-catalyzed addition and the oxymercuration–reduction process. The reaction was first reported by Herbert C. Brown in the late 1950s and it was recognized in his receiving the Nobel Prize in Chemistry in 1979.
In organic chemistry, hydroboration refers to the addition of a hydrogen-boron bond to certain double and triple bonds involving carbon. This chemical reaction is useful in the organic synthesis of organic compounds.

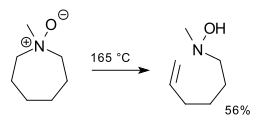
2,3-Sigmatropic rearrangements are a type of sigmatropic rearrangements and can be classified into two types. Rearrangements of allylic sulfoxides, amine oxides, selenoxides are neutral. Rearrangements of carbanions of allyl ethers are anionic. The general scheme for this kind of rearrangement is:

In chemistry, an amine oxide, also known as an amine N-oxide or simply N-oxide, is a chemical compound that has the chemical formula R3N+−O−. It contains a nitrogen-oxygen coordinate covalent bond with three additional hydrogen and/or substituent-groups attached to nitrogen. Sometimes it is written as R3N→O or, alternatively, as R3N=O.

In organic chemistry, aziridines are organic compounds containing the aziridine functional group, a three-membered heterocycle with one amine and two methylene bridges. The parent compound is aziridine, with molecular formula C2H4NH. Several drugs feature aziridine rings, including mitomycin C, porfiromycin, and azinomycin B (carzinophilin).
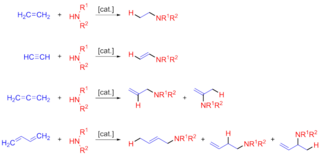
In organic chemistry, hydroamination is the addition of an N−H bond of an amine across a carbon-carbon multiple bond of an alkene, alkyne, diene, or allene. In the ideal case, hydroamination is atom economical and green. Amines are common in fine-chemical, pharmaceutical, and agricultural industries. Hydroamination can be used intramolecularly to create heterocycles or intermolecularly with a separate amine and unsaturated compound. The development of catalysts for hydroamination remains an active area, especially for alkenes. Although practical hydroamination reactions can be effected for dienes and electrophilic alkenes, the term hydroamination often implies reactions metal-catalyzed processes.
In organic chemistry, the Ei mechanism, also known as a thermal syn elimination or a pericyclic syn elimination, is a special type of elimination reaction in which two vicinal (adjacent) substituents on an alkane framework leave simultaneously via a cyclic transition state to form an alkene in a syn elimination. This type of elimination is unique because it is thermally activated and does not require additional reagents, unlike regular eliminations, which require an acid or base, or would in many cases involve charged intermediates. This reaction mechanism is often found in pyrolysis.
Selenoxide elimination is a method for the chemical synthesis of alkenes from selenoxides. It is most commonly used to synthesize α,β-unsaturated carbonyl compounds from the corresponding saturated analogues. It is mechanistically related to the Cope reaction.
Amide reduction is a reaction in organic synthesis where an amide is reduced to either an amine or an aldehyde functional group.
Electrophilic amination is a chemical process involving the formation of a carbon–nitrogen bond through the reaction of a nucleophilic carbanion with an electrophilic source of nitrogen.

An oxaziridine is an organic molecule that features a three-membered heterocycle containing oxygen, nitrogen, and carbon. In their largest application, oxaziridines are intermediates in the industrial production of hydrazine. Oxaziridine derivatives are also used as specialized reagents in organic chemistry for a variety of oxidations, including alpha hydroxylation of enolates, epoxidation and aziridination of olefins, and other heteroatom transfer reactions. Oxaziridines also serve as precursors to amides and participate in [3+2] cycloadditions with various heterocumulenes to form substituted five-membered heterocycles. Chiral oxaziridine derivatives effect asymmetric oxygen transfer to prochiral enolates as well as other substrates. Some oxaziridines also have the property of a high barrier to inversion of the nitrogen, allowing for the possibility of chirality at the nitrogen center.
The Saegusa–Ito oxidation is a chemical reaction used in organic chemistry. It was discovered in 1978 by Takeo Saegusa and Yoshihiko Ito as a method to introduce α-β unsaturation in carbonyl compounds. The reaction as originally reported involved formation of a silyl enol ether followed by treatment with palladium(II) acetate and benzoquinone to yield the corresponding enone. The original publication noted its utility for regeneration of unsaturation following 1,4-addition with nucleophiles such as organocuprates.
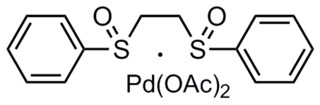
The White catalyst is a transition metal coordination complex named after the chemist by whom it was first synthesized, M. Christina White, a professor at the University of Illinois. The catalyst has been used in a variety of allylic C-H functionalization reactions of α-olefins. In addition, it has been shown to catalyze oxidative Heck reactions.
In chemistry, metal-catalysed hydroboration is a reaction used in organic synthesis. It is one of several examples of homogeneous catalysis.
A nitroalkene, or nitro olefin, is a functional group combining the functionality of its constituent parts, an alkene and nitro group, while displaying its own chemical properties through alkene activation, making the functional group useful in specialty reactions such as the Michael reaction or Diels-Alder additions.

Organotantalum chemistry is the chemistry of chemical compounds containing a carbon-to-tantalum chemical bond. A wide variety of compound have been reported, initially with cyclopentadienyl and CO ligands. Oxidation states vary from Ta(V) to Ta(-I).
The Riley oxidation is a selenium dioxide-mediated oxidation of methylene groups adjacent to carbonyls. It was first reported by Riley and co-workers in 1932. In the decade that ensued, selenium-mediated oxidation rapidly expanded in use, and in 1939, Guillemonat and co-workers disclosed the selenium dioxide-mediated oxidation of olefins at the allylic position. Today, selenium-dioxide-mediated oxidation of methylene groups to alpha ketones and at the allylic position of olefins is known as the Riley Oxidation.
In organic chemistry, the Davis oxidation or Davis' oxaziridine oxidation refers to oxidations involving the use of the Davis reagent or other similar oxaziridine reagents. This reaction mainly refers to the generation of α-hydroxy carbonyl compounds (acyloins) from ketones or esters. The reaction is carried out in a basic environment to generate the corresponding enolate from the ketone or ester. This reaction has been shown to work for amides.
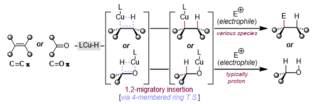
A hydrocupration is a chemical reaction whereby a ligated copper hydride species, reacts with a carbon-carbon or carbon-oxygen pi-system; this insertion is typically thought to occur via a four-membered ring transition state, producing a new copper-carbon or copper-oxygen sigma-bond and a stable (generally) carbon-hydrogen sigma-bond. In the latter instance (copper-oxygen), protonation (protodemetalation) is typical – the former (copper-carbon) has broad utility. The generated copper-carbon bond (organocuprate) has been employed in various nucleophilic additions to polar conjugated and non-conjugated systems and has also been used to forge new carbon-heteroatom bonds.