
In organic chemistry, an alkene, or olefin, is a hydrocarbon containing a carbon–carbon double bond. The double bond may be internal or in the terminal position. Terminal alkenes are also known as α-olefins.

In organic chemistry, allenes are organic compounds in which one carbon atom has double bonds with each of its two adjacent carbon atoms. Allenes are classified as cumulated dienes. The parent compound of this class is propadiene, which is itself also called allene. A group of the structure R2C=C=CR− is called allenyl, while a substituent attached to an allene is referred to as an allenic substituent. In analogy to allylic and propargylic, a substituent attached to a saturated carbon α to an allene is referred to as an allenylic substituent. While allenes have two consecutive ('cumulated') double bonds, compounds with three or more cumulated double bonds are called cumulenes.

In organic chemistry, a diene ; also diolefin, dy-OH-lə-fin) or alkadiene) is a covalent compound that contains two double bonds, usually among carbon atoms. They thus contain two alkene units, with the standard prefix di of systematic nomenclature. As a subunit of more complex molecules, dienes occur in naturally occurring and synthetic chemicals and are used in organic synthesis. Conjugated dienes are widely used as monomers in the polymer industry. Polyunsaturated fats are of interest to nutrition.

In organic chemistry, an acetal is a functional group with the connectivity R2C(OR')2. Here, the R groups can be organic fragments or hydrogen, while the R' groups must be organic fragments not hydrogen. The two R' groups can be equivalent to each other or not. Acetals are formed from and convertible to aldehydes or ketones and have the same oxidation state at the central carbon, but have substantially different chemical stability and reactivity as compared to the analogous carbonyl compounds. The central carbon atom has four bonds to it, and is therefore saturated and has tetrahedral geometry.
An ylide or ylid is a neutral dipolar molecule containing a formally negatively charged atom (usually a carbanion) directly attached to a heteroatom with a formal positive charge (usually nitrogen, phosphorus or sulfur), and in which both atoms have full octets of electrons. The result can be viewed as a structure in which two adjacent atoms are connected by both a covalent and an ionic bond; normally written X+–Y−. Ylides are thus 1,2-dipolar compounds, and a subclass of zwitterions. They appear in organic chemistry as reagents or reactive intermediates.

In organic chemistry, an imine is a functional group or organic compound containing a carbon–nitrogen double bond. The nitrogen atom can be attached to a hydrogen or an organic group (R). The carbon atom has two additional single bonds. Imines are common in synthetic and naturally occurring compounds and they participate in many reactions.

In organic chemistry, the ene reaction is a chemical reaction between an alkene with an allylic hydrogen and a compound containing a multiple bond, in order to form a new σ-bond with migration of the ene double bond and 1,5 hydrogen shift. The product is a substituted alkene with the double bond shifted to the allylic position.
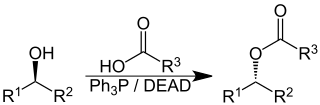
The Mitsunobu reaction is an organic reaction that converts an alcohol into a variety of functional groups, such as an ester, using triphenylphosphine and an azodicarboxylate such as diethyl azodicarboxylate (DEAD) or diisopropyl azodicarboxylate (DIAD). Although DEAD and DIAD are most commonly used, there are a variety of other azodicarboxylates available which facilitate an easier workup and/or purification and in some cases, facilitate the use of more basic nucleophiles. It was discovered by Oyo Mitsunobu (1934–2003). In a typical protocol, one dissolves the alcohol, the carboxylic acid, and triphenylphosphine in tetrahydrofuran or other suitable solvent, cool to 0 °C using an ice-bath, slowly add the DEAD dissolved in THF, then stir at room temperature for several hours. The alcohol reacts with the phosphine to create a good leaving group then undergoes an inversion of stereochemistry in classic SN2 fashion as the nucleophile displaces it. A common side-product is produced when the azodicarboxylate displaces the leaving group instead of the desired nucleophile. This happens if the nucleophile is not acidic enough or is not nucleophilic enough due to steric or electronic constraints. A variation of this reaction utilizing a nitrogen nucleophile is known as a Fukuyama–Mitsunobu.

The Claisen rearrangement is a powerful carbon–carbon bond-forming chemical reaction discovered by Rainer Ludwig Claisen. The heating of an allyl vinyl ether will initiate a [3,3]-sigmatropic rearrangement to give a γ,δ-unsaturated carbonyl, driven by exergonically favored carbonyl CO bond formation (Δ = −327 kcal/mol.
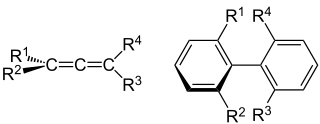
In chemistry, axial chirality is a special case of chirality in which a molecule contains two pairs of chemical groups in a non-planar arrangement about an axis of chirality so that the molecule is not superposable on its mirror image. The axis of chirality is usually determined by a chemical bond that is constrained against free rotation either by steric hindrance of the groups, as in substituted biaryl compounds such as BINAP, or by torsional stiffness of the bonds, as in the C=C double bonds in allenes such as glutinic acid. Axial chirality is most commonly observed in substituted biaryl compounds wherein the rotation about the aryl–aryl bond is restricted so it results in chiral atropisomers, as in various ortho-substituted biphenyls, and in binaphthyls such as BINAP.
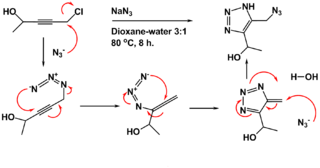
The Banert cascade is an organic reaction in which an NH-1,2,3-triazole is prepared from a propargyl halide or sulfate and sodium azide in a dioxane- water mixture at elevated temperatures. It is named after Klaus Banert, who first reported the process in 1989. This cascade reaction is unusual because it consists of two consecutive rearrangement reactions.
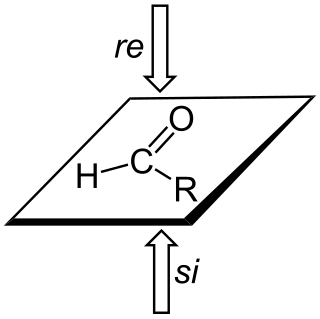
In stereochemistry, prochiral molecules are those that can be converted from achiral to chiral in a single step. An achiral species which can be converted to a chiral in two steps is called proprochiral.

A cumulene is a compound having three or more cumulative (consecutive) double bonds. They are analogous to allenes, only having a more extensive chain. The simplest molecule in this class is butatriene, which is also called simply cumulene. Unlike most alkanes and alkenes, cumulenes tend to be rigid, comparable to polyynes. Cumulene carbenes H2Cn for n from 3 to 6 have been observed in interstellar molecular clouds and in laboratory experiments by using microwave and infrared spectroscopy. Cumulenes containing heteroatoms are called heterocumulenes; an example is carbon suboxide.
A Norrish reaction, named after Ronald George Wreyford Norrish, is a photochemical reaction taking place with ketones and aldehydes. Such reactions are subdivided into Norrish type I reactions and Norrish type II reactions. While of limited synthetic utility these reactions are important in the photo-oxidation of polymers such as polyolefins, polyesters, certain polycarbonates and polyketones.

In organic chemistry, cyclopropanation refers to any chemical process which generates cyclopropane rings. It is an important process in modern chemistry as many useful compounds bear this motif; for example pyrethroid insecticides and a number of quinolone antibiotics. However, the high ring strain present in cyclopropanes makes them challenging to produce and generally requires the use of highly reactive species, such as carbenes, ylids and carbanions. Many of the reactions proceed in a cheletropic manner.
The [2,3]-Wittig rearrangement is the transformation of an allylic ether into a homoallylic alcohol via a concerted, pericyclic process. Because the reaction is concerted, it exhibits a high degree of stereocontrol, and can be employed early in a synthetic route to establish stereochemistry. The Wittig rearrangement requires strongly basic conditions, however, as a carbanion intermediate is essential. [1,2]-Wittig rearrangement is a competitive process.
The Kharasch–Sosnovsky reaction is a method that involves using a copper or cobalt salt as a catalyst to oxidize olefins at the allylic position, subsequently condensing a peroxy ester or a peroxide resulting in the formation of allylic benzoates or alcohols via radical oxidation. This method is noteworthy for being the first allylic functionalization to utilize first-row transition metals and has found numerous applications in chemical and total synthesis. Chiral ligands can be used to render the reaction asymmetric, constructing chiral C–O bonds via C–H bond activation. This is notable as asymmetric addition to allylic groups tends to be difficult due to the transition state being highly symmetric. The reaction is named after Morris S. Kharasch and George Sosnovsky who first reported it in 1958. This method is noteworthy for being the first allylic functionalization to utilize first-row transition metals and has found numerous applications in chemical and total synthesis.
The metallo-ene reaction is a chemical reaction employed within organic synthesis. Mechanistically similar to the classic ene reaction, the metallo-ene reaction involves a six-member cyclic transition state that brings an allylic species and an alkene species together to undergo a rearrangement. The initial allylic group migrates to one terminus of the alkene reactant and a new carbon-carbon sigma bond is formed between the allylic species and the other terminus of the alkene reactant. In the metallo-ene reaction, a metal ion acts as the migrating group rather than a hydrogen atom as in the classic ene reaction.
The Crabbé reaction is an organic reaction that converts a terminal alkyne and aldehyde into an allene in the presence of a soft Lewis acid catalyst and secondary amine. Given continued developments in scope and generality, it is a convenient and increasingly important method for the preparation of allenes, a class of compounds often viewed as exotic and synthetically challenging to access.
In organic chemistry, the Myers deoxygenation reaction is an organic redox reaction that reduces an alcohol into an alkyl position by way of an arenesulfonylhydrazine as a key intermediate. This name reaction is one of four discovered by Andrew Myers that are named after him; this reaction and the Myers allene synthesis reaction involve the same type of intermediate. The other reactions are Myers' asymmetric alkylation and Myers-Saito Cycloaromatization.
