Related Research Articles

In organic chemistry, an alkene is a hydrocarbon containing a carbon–carbon double bond. The double bond may be internal or in the terminal position. Terminal alkenes are also known as α-olefins.

In organic chemistry, allenes are organic compounds in which one carbon atom has double bonds with each of its two adjacent carbon atoms. Allenes are classified as cumulated dienes. The parent compound of this class is propadiene, which is itself also called allene. An group of the structure R2C=C=CR− is called allenyl, where R is H or some alkyl group. Compounds with an allene-type structure but with more than three carbon atoms are members of a larger class of compounds called cumulenes with X=C=Y bonding.
The Wolff–Kishner reduction is a reaction used in organic chemistry to convert carbonyl functionalities into methylene groups. In the context of complex molecule synthesis, it is most frequently employed to remove a carbonyl group after it has served its synthetic purpose of activating an intermediate in a preceding step. As such, there is no obvious retron for this reaction. The reaction was reported by Nikolai Kischner in 1911 and Ludwig Wolff in 1912.

The Michaelis–Arbuzov reaction is the chemical reaction of a trivalent phosphorus ester with an alkyl halide to form a pentavalent phosphorus species and another alkyl halide. The picture below shows the most common types of substrates undergoing the Arbuzov reaction; phosphite esters (1) react to form phosphonates (2), phosphonites (3) react to form phosphinates (4) and phosphinites (5) react to form phosphine oxides (6).

The Bamford–Stevens reaction is a chemical reaction whereby treatment of tosylhydrazones with strong base gives alkenes. It is named for the British chemist William Randall Bamford and the Scottish chemist Thomas Stevens Stevens (1900–2000). The usage of aprotic solvents gives predominantly Z-alkenes, while protic solvent gives a mixture of E- and Z-alkenes. As an alkene-generating transformation, the Bamford–Stevens reaction has broad utility in synthetic methodology and complex molecule synthesis.

The Curtius rearrangement, first defined by Theodor Curtius in 1885, is the thermal decomposition of an acyl azide to an isocyanate with loss of nitrogen gas. The isocyanate then undergoes attack by a variety of nucleophiles such as water, alcohols and amines, to yield a primary amine, carbamate or urea derivative respectively. Several reviews have been published.
The Barton–McCombie deoxygenation is an organic reaction in which a hydroxy functional group in an organic compound is replaced by a hydrogen to give an alkyl group. It is named after British chemists Sir Derek Harold Richard Barton and Stuart W. McCombie.
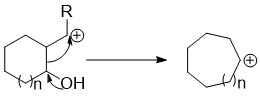
Ring expansion and ring contraction reactions expand or contract rings, usually in organic chemistry. The term usually refers to reactions involve making and breaking C-C bonds, Diverse mechanisms lead to these kinds of reactions.

The Wolff rearrangement is a reaction in organic chemistry in which an α-diazocarbonyl compound is converted into a ketene by loss of dinitrogen with accompanying 1,2-rearrangement. The Wolff rearrangement yields a ketene as an intermediate product, which can undergo nucleophilic attack with weakly acidic nucleophiles such as water, alcohols, and amines, to generate carboxylic acid derivatives or undergo [2+2] cycloaddition reactions to form four-membered rings. The mechanism of the Wolff rearrangement has been the subject of debate since its first use. No single mechanism sufficiently describes the reaction, and there are often competing concerted and carbene-mediated pathways; for simplicity, only the textbook, concerted mechanism is shown below. The reaction was discovered by Ludwig Wolff in 1902. The Wolff rearrangement has great synthetic utility due to the accessibility of α-diazocarbonyl compounds, variety of reactions from the ketene intermediate, and stereochemical retention of the migrating group. However, the Wolff rearrangement has limitations due to the highly reactive nature of α-diazocarbonyl compounds, which can undergo a variety of competing reactions.

Organozinc chemistry is the study of the physical properties, synthesis, and reactions of organozinc compounds, which are organometallic compounds that contain carbon (C) to zinc (Zn) chemical bonds.
The Barton reaction, also known as the Barton nitrite ester reaction, is a photochemical reaction that involves the photolysis of an alkyl nitrite to form a δ-nitroso alcohol.
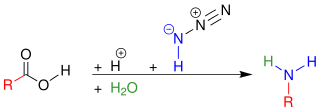
In organic chemistry, the Schmidt reaction is an organic reaction in which an azide reacts with a carbonyl derivative, usually an aldehyde, ketone, or carboxylic acid, under acidic conditions to give an amine or amide, with expulsion of nitrogen. It is named after Karl Friedrich Schmidt (1887–1971), who first reported it in 1924 by successfully converting benzophenone and hydrazoic acid to benzanilide. The intramolecular reaction was not reported until 1991 but has become important in the synthesis of natural products. The reaction is effective with carboxylic acids to give amines (above), and with ketones to give amides (below).
The Markó–Lam deoxygenation is an organic chemistry reaction where the hydroxy functional group in an organic compound is replaced by a hydrogen atom to give an alkyl group. The Markó-Lam reaction is a variant of the Bouveault–Blanc reduction and an alternative to the classical Barton–McCombie deoxygenation. It is named for the Belgian chemists István Markó and Kevin Lam.

Reductions with samarium(II) iodide involve the conversion of various classes of organic compounds into reduced products through the action of samarium(II) iodide, a mild one-electron reducing agent.
Desulfonylation reactions are chemical reactions leading to the removal of a sulfonyl group from organic compounds. As the sulfonyl functional group is electron-withdrawing, methods for cleaving the sulfur–carbon bonds of sulfones are typically reductive in nature. Olefination or replacement with hydrogen may be accomplished using reductive desulfonylation methods.
The Mislow–Evans rearrangement is a name reaction in organic chemistry. It is named after Kurt Mislow who reported the prototypical reaction in 1966, and David A. Evans who published further developments. The reaction allows the formation of allylic alcohols from allylic sulfoxides in a 2,3-sigmatropic rearrangement.

Trifluoroperacetic acid is an organofluorine compound, the peroxy acid analog of trifluoroacetic acid, with the condensed structural formula CF
3COOOH. It is a strong oxidizing agent for organic oxidation reactions, such as in Baeyer–Villiger oxidations of ketones. It is the most reactive of the organic peroxy acids, allowing it to successfully oxidise relatively unreactive alkenes to epoxides where other peroxy acids are ineffective. It can also oxidise the chalcogens in some functional groups, such as by transforming selenoethers to selones. It is a potentially explosive material and is not commercially available, but it can be quickly prepared as needed. Its use as a laboratory reagent was pioneered and developed by William D. Emmons.

Bis(cyclopentadienyl)titanium(III) chloride, also known as the Nugent–RajanBabu reagent, is the organotitanium compound which exists as a dimer with the formula [(C5H5)2TiCl]2. It is an air sensitive green solid. The complex finds specialized use in synthetic organic chemistry as a single electron reductant.
Vinylcyclopropane [5+2] cycloaddition is a type of cycloaddition between a vinylcyclopropane (VCP) and an olefin or alkyne to form a seven-membered ring.
In organic chemistry, the Myers allene synthesis is a chemical reaction that converts a propargyl alcohol into an allene by way of an arenesulfonylhydrazine as a key intermediate. This name reaction is one of two discovered by Andrew Myers that are named after him; both this reaction and the Myers deoxygenation reaction involve the same type of intermediate.
References
- 1 2 3 Li, Jie Jack, ed. (2009). "6.3.5. Miscellaneous Synthetic Utility—Allene synthesis". Name Reactions for Homologations. Vol. Part 2. Wiley. pp. 727–728. doi:10.1002/9780470487044. ISBN 9780470487044..
- ↑ Kürti, László; Czakó, Barbara (29 September 2005). Strategic Applications of Named reactions in Organic Synthesis. Elsevier. pp. 300–301. ISBN 0124297854.
- ↑ Hassner, Alfred; Namboothiri, Irishi (2012). Organic Synthesis Based Name Reactions third edition. Elsevier. p. 333. doi:10.1016/C2009-0-30489-4. ISBN 9780080966304.
- 1 2 Myers, Andrew G.; Movassaghi, Mohammad; Zheng, Bin (1997). "Single-Step Process for the Reductive Deoxygenation of Unhindered Alcohols". J. Am. Chem. Soc. 119 (36): 8572–8573. doi:10.1021/ja971768v.
| | This organic chemistry article is a stub. You can help Wikipedia by expanding it. |