In organic chemistry, a cycloaddition is a chemical reaction in which "two or more unsaturated molecules combine with the formation of a cyclic adduct in which there is a net reduction of the bond multiplicity". The resulting reaction is a cyclization reaction. Many but not all cycloadditions are concerted and thus pericyclic. Nonconcerted cycloadditions are not pericyclic. As a class of addition reaction, cycloadditions permit carbon–carbon bond formation without the use of a nucleophile or electrophile.
The 1,3-dipolar cycloaddition is a chemical reaction between a 1,3-dipole and a dipolarophile to form a five-membered ring. The earliest 1,3-dipolar cycloadditions were described in the late 19th century to the early 20th century, following the discovery of 1,3-dipoles. Mechanistic investigation and synthetic application were established in the 1960s, primarily through the work of Rolf Huisgen. Hence, the reaction is sometimes referred to as the Huisgen cycloaddition. 1,3-dipolar cycloaddition is an important route to the regio- and stereoselective synthesis of five-membered heterocycles and their ring-opened acyclic derivatives. The dipolarophile is typically an alkene or alkyne, but can be other pi systems. When the dipolarophile is an alkyne, aromatic rings are generally produced.
An alkyne trimerisation is a [2+2+2] cycloaddition reaction in which three alkyne units react to form a benzene ring. The reaction requires a metal catalyst. The process is of historic interest as well as being applicable to organic synthesis. Being a cycloaddition reaction, it has high atom economy. Many variations have been developed, including cyclisation of mixtures of alkynes and alkenes as well as alkynes and nitriles.

The Pauson–Khand (PK) reaction is a chemical reaction, described as a [2+2+1] cycloaddition. In it, an alkyne, an alkene and carbon monoxide combine into a α,β-cyclopentenone in the presence of a metal-carbonyl catalyst.
The Paternò–Büchi reaction, named after Emanuele Paternò and George Büchi, who established its basic utility and form, is a photochemical reaction, specifically a 2+2 photocycloaddition, which forms four-membered oxetane rings from an excited carbonyl and reacting with an alkene.
Ring-closing metathesis (RCM) is a widely used variation of olefin metathesis in organic chemistry for the synthesis of various unsaturated rings via the intramolecular metathesis of two terminal alkenes, which forms the cycloalkene as the E- or Z- isomers and volatile ethylene.

Azomethine ylides are nitrogen-based 1,3-dipoles, consisting of an iminium ion next to a carbanion. They are used in 1,3-dipolar cycloaddition reactions to form five-membered heterocycles, including pyrrolidines and pyrrolines. These reactions are highly stereo- and regioselective, and have the potential to form four new contiguous stereocenters. Azomethine ylides thus have high utility in total synthesis, and formation of chiral ligands and pharmaceuticals. Azomethine ylides can be generated from many sources, including aziridines, imines, and iminiums. They are often generated in situ, and immediately reacted with dipolarophiles.
In organic chemistry the Brook rearrangement refers to any [1,n] carbon to oxygen silyl migration. The rearrangement was first observed in the late 1950s by Canadian chemist Adrian Gibbs Brook (1924–2013), after which the reaction is named. These migrations can be promoted in a number of different ways, including thermally, photolytically or under basic/acidic conditions. In the forward direction, these silyl migrations produce silyl ethers as products which is driven by the stability of the oxygen-silicon bond.
The Stetter reaction is a reaction used in organic chemistry to form carbon-carbon bonds through a 1,4-addition reaction utilizing a nucleophilic catalyst. While the related 1,2-addition reaction, the benzoin condensation, was known since the 1830s, the Stetter reaction was not reported until 1973 by Dr. Hermann Stetter. The reaction provides synthetically useful 1,4-dicarbonyl compounds and related derivatives from aldehydes and Michael acceptors. Unlike 1,3-dicarbonyls, which are easily accessed through the Claisen condensation, or 1,5-dicarbonyls, which are commonly made using a Michael reaction, 1,4-dicarbonyls are challenging substrates to synthesize, yet are valuable starting materials for several organic transformations, including the Paal–Knorr synthesis of furans and pyrroles. Traditionally utilized catalysts for the Stetter reaction are thiazolium salts and cyanide anion, but more recent work toward the asymmetric Stetter reaction has found triazolium salts to be effective. The Stetter reaction is an example of umpolung chemistry, as the inherent polarity of the aldehyde is reversed by the addition of the catalyst to the aldehyde, rendering the carbon center nucleophilic rather than electrophilic.
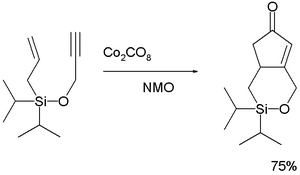
The Nazarov cyclization reaction is a chemical reaction used in organic chemistry for the synthesis of cyclopentenones. The reaction is typically divided into classical and modern variants, depending on the reagents and substrates employed. It was originally discovered by Ivan Nikolaevich Nazarov (1906–1957) in 1941 while studying the rearrangements of allyl vinyl ketones.
In organosilicon chemistry, silyl enol ethers are a class of organic compounds that share the common functional group R3Si−O−CR=CR2, composed of an enolate bonded to a silane through its oxygen end and an ethene group as its carbon end. They are important intermediates in organic synthesis.
In organic chemistry, an intramolecular Diels-Alder cycloaddition is a Diels–Alder reaction in which the diene and the dienophile are both part of the same molecule. The reaction leads to the formation of the cyclohexene-like structure as usual for a Diels–Alder reaction, but as part of a more complex fused or bridged cyclic ring system. This reaction can gives rise to various natural derivatives of decalin.
Trimethylenemethane cycloaddition is the formal [3+2] annulation of trimethylenemethane (TMM) derivatives to two-atom pi systems. Although TMM itself is too reactive and unstable to be stored, reagents which can generate TMM or TMM synthons in situ can be used to effect cycloaddition reactions with appropriate electron acceptors. Generally, electron-deficient pi bonds undergo cyclization with TMMs more easily than electron-rich pi bonds.
In organic chemistry, enone–alkene cycloadditions are a version of the [2+2] cycloaddition This reaction involves an enone and alkene as substrates. Although the concerted photochemical [2+2] cycloaddition is allowed, the reaction between enones and alkenes is stepwise and involves discrete diradical intermediates.
The nitrone-olefin [3+2] cycloaddition reaction is the combination of a nitrone with an alkene or alkyne to generate an isoxazoline or isoxazolidine via a [3+2] cycloaddition process. This reaction is a 1,3-dipolar cycloaddition, in which the nitrone acts as the 1,3-dipole, and the alkene or alkyne as the dipolarophile.
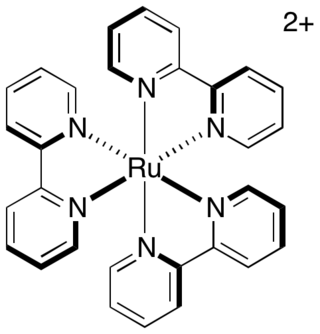
Photoredox catalysis is a branch of photochemistry that uses single-electron transfer. Photoredox catalysts are generally drawn from three classes of materials: transition-metal complexes, organic dyes, and semiconductors. While organic photoredox catalysts were dominant throughout the 1990s and early 2000s, soluble transition-metal complexes are more commonly used today.

Alkene carboamination is the simultaneous formation of C–N and C–C bonds across an alkene. This method represents a powerful strategy to build molecular complexity with up to two stereocenters in a single operation. Generally, there are four categories of reaction modes for alkene carboamination. The first class is cyclization reactions, which will form a N-heterocycle as a result. The second class has been well established in the last decade. Alkene substrates with a tethered nitrogen nucleophile have been used in these transformations to promote intramolecular aminocyclization. While intermolecular carboamination is extremely hard, people have developed a strategy to combine the nitrogen and carbon part, which is known as the third class. The most general carboamination, which takes three individual parts and couples them together is still underdeveloped.

In organometallic chemistry, the activation of cyclopropanes by transition metals is a research theme with implications for organic synthesis and homogeneous catalysis. Being highly strained, cyclopropanes are prone to oxidative addition to transition metal complexes. The resulting metallacycles are susceptible to a variety of reactions. These reactions are rare examples of C-C bond activation. The rarity of C-C activation processes has been attributed to Steric effects that protect C-C bonds. Furthermore, the directionality of C-C bonds as compared to C-H bonds makes orbital interaction with transition metals less favorable. Thermodynamically, C-C bond activation is more favored than C-H bond activation as the strength of a typical C-C bond is around 90 kcal per mole while the strength of a typical unactivated C-H bond is around 104 kcal per mole.
Carbonyl olefin metathesis is a type of metathesis reaction that entails, formally, the redistribution of fragments of an alkene and a carbonyl by the scission and regeneration of carbon-carbon and carbon-oxygen double bonds respectively. It is a powerful method in organic synthesis using simple carbonyls and olefins and converting them into less accessible products with higher structural complexity.
In organic chemistry, the Conia-ene reaction is an intramolecular cyclization reaction between an enolizable carbonyl such as an ester or ketone and an alkyne or alkene, giving a cyclic product with a new carbon-carbon bond. As initially reported by J. M. Conia and P. Le Perchec, the Conia-ene reaction is a heteroatom analog of the ene reaction that uses an enol as the ene component. Like other pericyclic reactions, the original Conia-ene reaction required high temperatures to proceed, limiting its wider application. However, subsequent improvements, particularly in metal catalysis, have led to significant expansion of reaction scope. Consequently, various forms of the Conia-ene reaction have been employed in the synthesis of complex molecules and natural products.



![Tethered intramolecular [2+2] reactions 23 fig. 1.png](http://upload.wikimedia.org/wikipedia/commons/8/88/23_fig._1.png)


![Effects of the length of tether on [2+2] photocyclization reaction 23 fig. 2.png](http://upload.wikimedia.org/wikipedia/commons/8/82/23_fig._2.png)
![Tethered [2+2] reaction in the total synthesis of (
+)
- Ginkgolide B 23 fig. 3.png](http://upload.wikimedia.org/wikipedia/commons/1/14/23_fig._3.png)