Cycloadditions can be described using two systems of notation. An older but still common notation is based on the size of linear arrangements of atoms in the reactants. It uses parentheses: (i + j + …) where the variables are the numbers of linear atoms in each reactant. The product is a cycle of size (i + j + …). In this system, the standard Diels-Alder reaction is a [4+2]-cycloaddition, the 1,3-dipolar cycloaddition is a [3+2]-cycloaddition and cyclopropanation of a carbene with an alkene a [2+1]-cycloaddition.[1]
A more recent, IUPAC-preferred notation, first introduced by Woodward and Hoffmann, uses square brackets to indicate the number of electrons, rather than carbon atoms, involved in the formation of the product. In the [i + j + ...] notation, the standard Diels-Alder reaction is a [4+2]-cycloaddition, while the 1,3-dipolar cycloaddition is also a [4+2]-cycloaddition.[1]
Thermal cycloadditions and their stereochemistry
Thermal cycloadditions are those cycloadditions where the reactants are in the ground electronic state. They usually have (4n + 2) π electrons participating in the starting material, for some integer n. These reactions occur for reasons of orbital symmetry in a suprafacial-suprafacial (syn/syn stereochemistry) in most cases. Very few examples of antarafacial-antarafacial (anti/anti stereochemistry) reactions have also been reported. There are a few examples of thermal cycloadditions which have 4n π electrons (for example the [2+2]-cycloaddition). These proceed in a suprafacial-antarafacial sense (syn/anti stereochemistry), such as the cycloaddition reactions of ketene and allene derivatives, in which the orthogonal set of p orbitals allows the reaction to proceed via a crossed transition state. Due to the involvement of an orthogonal orbital, this is classified as a pseudopericyclic reaction, to which the Woodward-Hoffmann rules do not apply to. Strained alkenes like trans-cycloheptene derivatives have also been reported to react in an antarafacial manner in [2+2]-cycloaddition reactions.
Doering (in a personal communication to Woodward) reported that heptafulvalene and tetracyanoethylene can react in a suprafacial-antarafacial [14+2]-cycloaddition. However, this reaction was later found to be stepwise, as it also produced the Woodward-Hoffmann forbidden suprafacial-suprafacial product under kinetic conditions. [3]
Erden and Kaufmann had previously found that the cycloaddition of heptafulvalene and N-phenyltriazolinedione also gave both suprafacial-antarafacial and suprafacial-suprafacial products, consistent with the stepwise nature of the analogous tetracyanoethylene reaction.[4]
Photochemical cycloadditions and their stereochemistry
Cycloadditions in which 4n π electrons participate can also occur via photochemical activation. Here, one component has an electron promoted from the HOMO (π bonding) to the LUMO (π* antibonding). Orbital symmetry is then such that the reaction can proceed in a suprafacial-suprafacial manner. An example is the DeMayo reaction. Another example is shown below, the photochemical dimerization of cinnamic acid.[5] The two transalkenes react head-to-tail, and the isolated isomers are called truxillic acids.
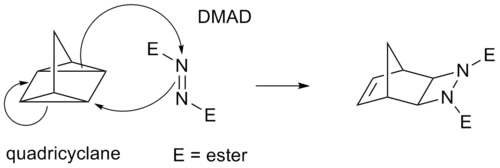
Some cycloadditions instead of π bonds operate through strained cyclopropane rings, as these have significant π character. For example, an analog for the Diels-Alder reaction is the quadricyclane-DMAD reaction:
In the (i+j+...) cycloaddition notation i and j refer to the number of atoms involved in the cycloaddition. In this notation, a Diels-Alder reaction is a [4+2]-cycloaddition and a 1,3-dipolar addition such as the first step in ozonolysis is a [3+2]-cycloaddition. The IUPAC preferred notation however, with [i+j+...] takes electrons into account and not atoms. In this notation, the DA reaction and the dipolar reaction both become a [4+2]-cycloaddition. The reaction between norbornadiene and an activated alkyne is a [2+2+2]-cycloaddition.
Cheletropic reactions are a subclass of cycloadditions. The key distinguishing feature of cheletropic reactions is that on one of the reagents, both new bonds are being made to the same atom. The classic example is the reaction of sulfur dioxide with a diene.
Cycloadditions often have metal-catalyzed and stepwise radical analogs, however these are not strictly speaking pericyclic reactions. When in a cycloaddition charged or radical intermediates are involved or when the cycloaddition result is obtained in a series of reaction steps they are sometimes called formal cycloadditions to make the distinction with true pericyclic cycloadditions.
An intermolecular formal [3+3] cycloaddition between an cyclic iminium chloride and cyclopentenone.
Iron-catalyzed [2+2] olefin cycloaddition
Iron[pyridine(diimine)] catalysts contain a redox active ligand in which the central iron atom can coordinate with two simple, unfunctionalized olefin double bonds. The catalyst can be written as a resonance between a structure containing unpaired electrons with the central iron atom in the II oxidation state, and one in which the iron is in the 0 oxidation state. This gives it the flexibility to engage in binding the double bonds as they undergo a cyclization reaction, generating a cyclobutane structure via C-C reductive elimination; alternatively a cyclobutene structure may be produced by beta-hydrogen elimination. Efficiency of the reaction varies substantially depending on the alkenes used, but rational ligand design may permit expansion of the range of reactions that can be catalyzed.[8][9]
↑Erden, Ihsan; KauFmann, Dieter (1981-01-01). "Cycloadditionsreaktionen des heptafulvalens". Tetrahedron Letters (in German). 22 (3): 215–218. doi:10.1016/0040-4039(81)80058-5. ISSN0040-4039.
↑Hein, Sara M. (June 2006). "An Exploration of a Photochemical Pericyclic Reaction Using NMR Data". Journal of Chemical Education. 83 (6): 940–942. Bibcode:2006JChEd..83..940H. doi:10.1021/ed083p940.
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.








![An intermolecular formal [3+3] cycloaddition between an cyclic iminium chloride and cyclopentenone. 3+3 cycloaddition - cyclic iminium to cyclic enone.svg](http://upload.wikimedia.org/wikipedia/commons/thumb/5/53/3%2B3_cycloaddition_-_cyclic_iminium_to_cyclic_enone.svg/500px-3%2B3_cycloaddition_-_cyclic_iminium_to_cyclic_enone.svg.png)