In a chemical reaction, chemical equilibrium is the state in which both the reactants and products are present in concentrations which have no further tendency to change with time, so that there is no observable change in the properties of the system. This state results when the forward reaction proceeds at the same rate as the reverse reaction. The reaction rates of the forward and backward reactions are generally not zero, but they are equal. Thus, there are no net changes in the concentrations of the reactants and products. Such a state is known as dynamic equilibrium.

A chemical reaction is a process that leads to the chemical transformation of one set of chemical substances to another. Classically, chemical reactions encompass changes that only involve the positions of electrons in the forming and breaking of chemical bonds between atoms, with no change to the nuclei, and can often be described by a chemical equation. Nuclear chemistry is a sub-discipline of chemistry that involves the chemical reactions of unstable and radioactive elements where both electronic and nuclear changes can occur.

In the Arrhenius model of reaction rates, activation energy is the minimum amount of energy that must be available to reactants for a chemical reaction to occur. The activation energy (Ea) of a reaction is measured in kilojoules per mole (kJ/mol) or kilocalories per mole (kcal/mol). Activation energy can be thought of as the magnitude of the potential barrier (sometimes called the energy barrier) separating minima of the potential energy surface pertaining to the initial and final thermodynamic state. For a chemical reaction to proceed at a reasonable rate, the temperature of the system should be high enough such that there exists an appreciable number of molecules with translational energy equal to or greater than the activation energy. The term "activation energy" was introduced in 1889 by the Swedish scientist Svante Arrhenius.

The reaction rate or rate of reaction is the speed at which a chemical reaction takes place, defined as proportional to the increase in the concentration of a product per unit time and to the decrease in the concentration of a reactant per unit time. Reaction rates can vary dramatically. For example, the oxidative rusting of iron under Earth's atmosphere is a slow reaction that can take many years, but the combustion of cellulose in a fire is a reaction that takes place in fractions of a second. For most reactions, the rate decreases as the reaction proceeds. A reaction's rate can be determined by measuring the changes in concentration over time.
Chemical kinetics, also known as reaction kinetics, is the branch of physical chemistry that is concerned with understanding the rates of chemical reactions. It is different from chemical thermodynamics, which deals with the direction in which a reaction occurs but in itself tells nothing about its rate. Chemical kinetics includes investigations of how experimental conditions influence the speed of a chemical reaction and yield information about the reaction's mechanism and transition states, as well as the construction of mathematical models that also can describe the characteristics of a chemical reaction.
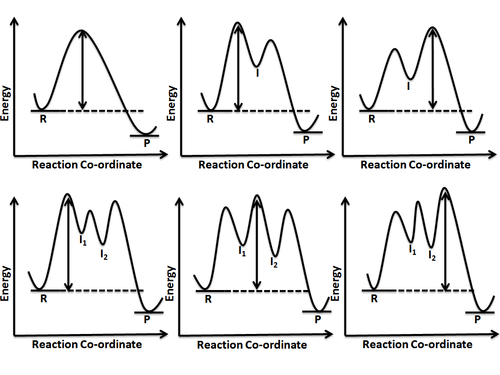
In chemistry, a reaction mechanism is the step by step sequence of elementary reactions by which overall chemical reaction occurs.

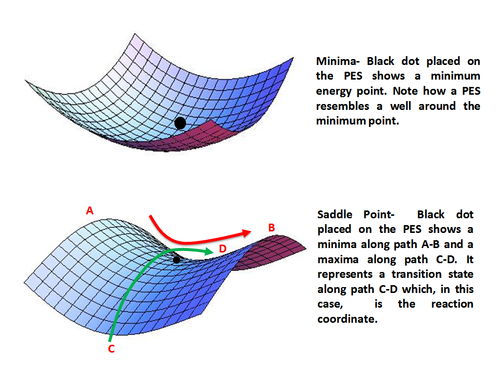
In chemistry, an activated complex represents a collection of intermediate structures in a chemical reaction when bonds are breaking and forming. The activated complex is an arrangement of atoms in an arbitrary region near the saddle point of a potential energy surface. The region represents not one defined state, but a range of unstable configurations that a collection of atoms pass through between the reactants and products of a reaction. Activated complexes have partial reactant and product character, which can significantly impact their behaviour in chemical reactions.

In chemistry, the transition state of a chemical reaction is a particular configuration along the reaction coordinate. It is defined as the state corresponding to the highest potential energy along this reaction coordinate. It is often marked with the double dagger (‡) symbol.
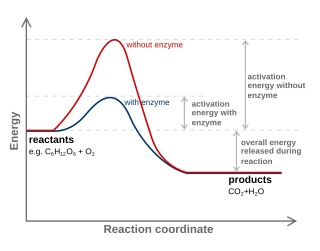
In chemistry, a reaction coordinate is an abstract one-dimensional coordinate chosen to represent progress along a reaction pathway. Where possible it is usually a geometric parameter that changes during the conversion of one or more molecular entities, such as bond length or bond angle. For example, in the homolytic dissociation of molecular hydrogen, an apt choice would be the coordinate corresponding to the bond length. Non-geometric parameters such as bond order are also used, but such direct representation of the reaction process can be difficult, especially for more complex reactions.
In chemical kinetics, the overall rate of a reaction is often approximately determined by the slowest step, known as the rate-determining step or rate-limiting step. For a given reaction mechanism, the prediction of the corresponding rate equation is often simplified by using this approximation of the rate-determining step.
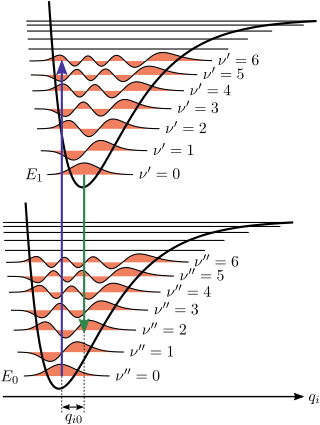
The Franck–Condon principle is a rule in spectroscopy and quantum chemistry that explains the intensity of vibronic transitions. The principle states that during an electronic transition, a change from one vibrational energy level to another will be more likely to happen if the two vibrational wave functions overlap more significantly.

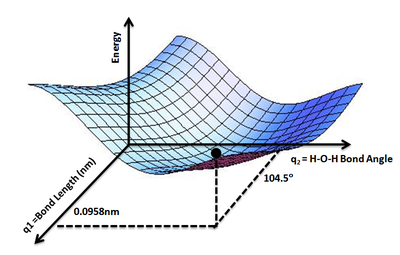
A potential energy surface (PES) or energy landscape describes the energy of a system, especially a collection of atoms, in terms of certain parameters, normally the positions of the atoms. The surface might define the energy as a function of one or more coordinates; if there is only one coordinate, the surface is called a potential energy curve or energy profile. An example is the Morse/Long-range potential.
In theoretical chemistry, Marcus theory is a theory originally developed by Rudolph A. Marcus, starting in 1956, to explain the rates of electron transfer reactions – the rate at which an electron can move or jump from one chemical species (called the electron donor) to another (called the electron acceptor). It was originally formulated to address outer sphere electron transfer reactions, in which the two chemical species only change in their charge with an electron jumping (e.g. the oxidation of an ion like Fe2+/Fe3+), but do not undergo large structural changes. It was extended to include inner sphere electron transfer contributions, in which a change of distances or geometry in the solvation or coordination shells of the two chemical species is taken into account (the Fe-O distances in Fe(H2O)2+ and Fe(H2O)3+ are different).
The Curtin–Hammett principle is a principle in chemical kinetics proposed by David Yarrow Curtin and Louis Plack Hammett. It states that, for a reaction that has a pair of reactive intermediates or reactants that interconvert rapidly, each going irreversibly to a different product, the product ratio will depend both on the difference in energy between the two conformers and the energy barriers from each of the rapidly equilibrating isomers to their respective products. Stated another way, the product distribution reflects the difference in energy between the two rate-limiting transition states. As a result, the product distribution will not necessarily reflect the equilibrium distribution of the two intermediates. The Curtin–Hammett principle has been invoked to explain selectivity in a variety of stereo- and regioselective reactions. The relationship between the (apparent) rate constants and equilibrium constant is known as the Winstein-Holness equation.

Hammond's postulate, is a hypothesis in physical organic chemistry which describes the geometric structure of the transition state in an organic chemical reaction. First proposed by George Hammond in 1955, the postulate states that:
If two states, as, for example, a transition state and an unstable intermediate, occur consecutively during a reaction process and have nearly the same energy content, their interconversion will involve only a small reorganization of the molecular structures.

In chemistry, transition state theory (TST) explains the reaction rates of elementary chemical reactions. The theory assumes a special type of chemical equilibrium (quasi-equilibrium) between reactants and activated transition state complexes.
Physical organic chemistry, a term coined by Louis Hammett in 1940, refers to a discipline of organic chemistry that focuses on the relationship between chemical structures and reactivity, in particular, applying experimental tools of physical chemistry to the study of organic molecules. Specific focal points of study include the rates of organic reactions, the relative chemical stabilities of the starting materials, reactive intermediates, transition states, and products of chemical reactions, and non-covalent aspects of solvation and molecular interactions that influence chemical reactivity. Such studies provide theoretical and practical frameworks to understand how changes in structure in solution or solid-state contexts impact reaction mechanism and rate for each organic reaction of interest.
More O’Ferrall–Jencks plots are two-dimensional representations of multiple reaction coordinate potential energy surfaces for chemical reactions that involve simultaneous changes in two bonds. As such, they are a useful tool to explain or predict how changes in the reactants or reaction conditions can affect the position and geometry of the transition state of a reaction for which there are possible competing pathways.
George Simms Hammond was an American scientist and theoretical chemist who developed "Hammond's postulate", and fathered organic photochemistry,–the general theory of the geometric structure of the transition state in an organic chemical reaction. Hammond's research is also known for its influence on the philosophy of science. His research garnered him the Norris Award in 1968, the Priestley Medal in 1976, the National Medal of Science in 1994, and the Othmer Gold Medal in 2003. He served as the executive chairman of the Allied Chemical Corporation from 1979 to 1989.
In chemistry, solvent effects are the influence of a solvent on chemical reactivity or molecular associations. Solvents can have an effect on solubility, stability and reaction rates and choosing the appropriate solvent allows for thermodynamic and kinetic control over a chemical reaction.
















![Figure 13: An energy profile diagram demonstrating the effect of a catalyst for the generic exothermic reaction of X + Y -Z. The catalyst offers an alternate reaction pathway (shown in red) where the rate determining step has a smaller DG[?]. The relative thermodynamic stabilities remain the same. Catalysis- Reaction progress.png](http://upload.wikimedia.org/wikipedia/commons/thumb/7/71/Catalysis-_Reaction_progress.png/310px-Catalysis-_Reaction_progress.png)