Overview
Theoretical chemistry unites principles and concepts common to all branches of chemistry. Within the framework of theoretical chemistry, there is a systematization of chemical laws, principles and rules, their refinement and detailing, the construction of a hierarchy. The central place in theoretical chemistry is occupied by the doctrine of the interconnection of the structure and properties of molecular systems. It uses mathematical and physical methods to explain the structures and dynamics of chemical systems and to correlate, understand, and predict their thermodynamic and kinetic properties. In the most general sense, it is explanation of chemical phenomena by methods of theoretical physics. In contrast to theoretical physics, in connection with the high complexity of chemical systems, theoretical chemistry, in addition to approximate mathematical methods, often uses semi-empirical and empirical methods.
In recent years, it has consisted primarily of quantum chemistry, i.e., the application of quantum mechanics to problems in chemistry. Other major components include molecular dynamics, statistical thermodynamics and theories of electrolyte solutions, reaction networks, polymerization, catalysis, molecular magnetism and spectroscopy.
Modern theoretical chemistry may be roughly divided into the study of chemical structure and the study of chemical dynamics. The former includes studies of: electronic structure, potential energy surfaces, and force fields; vibrational-rotational motion; equilibrium properties of condensed-phase systems and macro-molecules. Chemical dynamics includes: bimolecular kinetics and the collision theory of reactions and energy transfer; unimolecular rate theory and metastable states; condensed-phase and macromolecular aspects of dynamics.
Chemistry is the scientific study of the properties and behavior of matter. It is a physical science within the natural sciences that studies the chemical elements that make up matter and compounds made of atoms, molecules and ions: their composition, structure, properties, behavior and the changes they undergo during reactions with other substances. Chemistry also addresses the nature of chemical bonds in chemical compounds.

Computational chemistry is a branch of chemistry that uses computer simulations to assist in solving chemical problems. It uses methods of theoretical chemistry incorporated into computer programs to calculate the structures and properties of molecules, groups of molecules, and solids. The importance of this subject stems from the fact that, with the exception of some relatively recent findings related to the hydrogen molecular ion, achieving an accurate quantum mechanical depiction of chemical systems analytically, or in a closed form, is not feasible. The complexity inherent in the many-body problem exacerbates the challenge of providing detailed descriptions of quantum mechanical systems. While computational results normally complement information obtained by chemical experiments, it can occasionally predict unobserved chemical phenomena.
The following outline is provided as an overview of and topical guide to chemistry:

Physical chemistry is the study of macroscopic and microscopic phenomena in chemical systems in terms of the principles, practices, and concepts of physics such as motion, energy, force, time, thermodynamics, quantum chemistry, statistical mechanics, analytical dynamics and chemical equilibria.
Physical science is a branch of natural science that studies non-living systems, in contrast to life science. It in turn has many branches, each referred to as a "physical science", together is called the "physical sciences".
The following outline is provided as an overview of and topical guide to physics:
Quantum chemistry, also called molecular quantum mechanics, is a branch of physical chemistry focused on the application of quantum mechanics to chemical systems, particularly towards the quantum-mechanical calculation of electronic contributions to physical and chemical properties of molecules, materials, and solutions at the atomic level. These calculations include systematically applied approximations intended to make calculations computationally feasible while still capturing as much information about important contributions to the computed wave functions as well as to observable properties such as structures, spectra, and thermodynamic properties. Quantum chemistry is also concerned with the computation of quantum effects on molecular dynamics and chemical kinetics.
Atomic, molecular, and optical physics (AMO) is the study of matter–matter and light–matter interactions, at the scale of one or a few atoms and energy scales around several electron volts. The three areas are closely interrelated. AMO theory includes classical, semi-classical and quantum treatments. Typically, the theory and applications of emission, absorption, scattering of electromagnetic radiation (light) from excited atoms and molecules, analysis of spectroscopy, generation of lasers and masers, and the optical properties of matter in general, fall into these categories.

Molecular dynamics (MD) is a computer simulation method for analyzing the physical movements of atoms and molecules. The atoms and molecules are allowed to interact for a fixed period of time, giving a view of the dynamic "evolution" of the system. In the most common version, the trajectories of atoms and molecules are determined by numerically solving Newton's equations of motion for a system of interacting particles, where forces between the particles and their potential energies are often calculated using interatomic potentials or molecular mechanical force fields. The method is applied mostly in chemical physics, materials science, and biophysics.
Molecular physics is the study of the physical properties of molecules and molecular dynamics. The field overlaps significantly with physical chemistry, chemical physics, and quantum chemistry. It is often considered as a sub-field of atomic, molecular, and optical physics. Research groups studying molecular physics are typically designated as one of these other fields. Molecular physics addresses phenomena due to both molecular structure and individual atomic processes within molecules. Like atomic physics, it relies on a combination of classical and quantum mechanics to describe interactions between electromagnetic radiation and matter. Experiments in the field often rely heavily on techniques borrowed from atomic physics, such as spectroscopy and scattering.
Chemical physics is a branch of physics that studies chemical processes from a physical point of view. It focuses on understanding the physical properties and behavior of chemical systems, using principles from both physics and chemistry. This field investigates physicochemical phenomena using techniques from atomic and molecular physics and condensed matter physics.

Molecular modelling encompasses all methods, theoretical and computational, used to model or mimic the behaviour of molecules. The methods are used in the fields of computational chemistry, drug design, computational biology and materials science to study molecular systems ranging from small chemical systems to large biological molecules and material assemblies. The simplest calculations can be performed by hand, but inevitably computers are required to perform molecular modelling of any reasonably sized system. The common feature of molecular modelling methods is the atomistic level description of the molecular systems. This may include treating atoms as the smallest individual unit, or explicitly modelling protons and neutrons with its quarks, anti-quarks and gluons and electrons with its photons.

In the context of chemistry, molecular physics, physical chemistry, and molecular modelling, a force field is a computational model that is used to describe the forces between atoms within molecules or between molecules as well as in crystals. Force fields are a variety of interatomic potentials. More precisely, the force field refers to the functional form and parameter sets used to calculate the potential energy of a system on the atomistic level. Force fields are usually used in molecular dynamics or Monte Carlo simulations. The parameters for a chosen energy function may be derived from classical laboratory experiment data, calculations in quantum mechanics, or both. Force fields utilize the same concept as force fields in classical physics, with the main difference being that the force field parameters in chemistry describe the energy landscape on the atomistic level. From a force field, the acting forces on every particle are derived as a gradient of the potential energy with respect to the particle coordinates.
Car–Parrinello molecular dynamics or CPMD refers to either a method used in molecular dynamics or the computational chemistry software package used to implement this method.
Physical organic chemistry, a term coined by Louis Hammett in 1940, refers to a discipline of organic chemistry that focuses on the relationship between chemical structures and reactivity, in particular, applying experimental tools of physical chemistry to the study of organic molecules. Specific focal points of study include the rates of organic reactions, the relative chemical stabilities of the starting materials, reactive intermediates, transition states, and products of chemical reactions, and non-covalent aspects of solvation and molecular interactions that influence chemical reactivity. Such studies provide theoretical and practical frameworks to understand how changes in structure in solution or solid-state contexts impact reaction mechanism and rate for each organic reaction of interest.

Debashis Mukherjee is a theoretical chemist, well known for his research in the fields of molecular many body theory, theoretical spectroscopy, finite temperature non-perturbative many body theories. Mukherjee has been the first to develop and implement a class of many-body methods for electronic structure which are now standard works in the field. These methods, collectively called multireference coupled cluster formalisms, are versatile and powerful methods for predicting with quantitative accuracy the energetics and cross-sections of a vast range of molecular excitations and ionization. A long-standing problem of guaranteeing proper scaling of energy for many electron wave-functions of arbitrary complexity has also been first resolved by him. He has also been the first to develop a rigorously size-extensive state-specific multi-reference coupled cluster formalism, and its perturbative counterpart which is getting increasingly recognized as a very promising methodological advance.
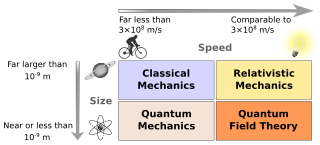
Physics is a scientific discipline that seeks to construct and experimentally test theories of the physical universe. These theories vary in their scope and can be organized into several distinct branches, which are outlined in this article.
The following outline is provided as an overview of and topical guide to natural science:
In computational chemistry, a solvent model is a computational method that accounts for the behavior of solvated condensed phases. Solvent models enable simulations and thermodynamic calculations applicable to reactions and processes which take place in solution. These include biological, chemical and environmental processes. Such calculations can lead to new predictions about the physical processes occurring by improved understanding.
