Shown here is a bioorthogonal ligation between biomolecule X and reactive partner Y. To be considered bioorthogonal, these reactive partners cannot perturb other chemical functionality naturally found within the cell.
The use of bioorthogonal chemistry typically proceeds in two steps. First, a cellular substrate is modified with a bioorthogonal functional group (chemical reporter) and introduced to the cell; substrates include metabolites, enzyme inhibitors, etc. The chemical reporter must not alter the structure of the substrate dramatically to avoid affecting its bioactivity. Secondly, a probe containing the complementary functional group is introduced to react and label the substrate.
Although effective bioorthogonal reactions such as copper-free click chemistry have been developed, development of new reactions continues to generate orthogonal methods for labeling to allow multiple methods of labeling to be used in the same biosystems. Carolyn R. Bertozzi was awarded the Nobel Prize in Chemistry in 2022 for her development of click chemistry and bioorthogonal chemistry.[14]
Etymology
The word bioorthogonal comes from Greek bio- "living" and orthogōnios "right-angled". Thus literally a reaction that goes perpendicular to a living system, thus not disturbing it.
Requirements for bioorthogonality
To be considered bioorthogonal, a reaction must fulfill a number of requirements:
Selectivity: The reaction must be selective between endogenous functional groups to avoid side reactions with biological compounds
Biological inertness: Reactive partners and resulting linkage should not possess any mode of reactivity capable of disrupting the native chemical functionality of the organism under study.
Chemical inertness: The covalent link should be strong and inert to biological reactions.
Kinetics: The reaction must be rapid so that covalent ligation is achieved prior to probe metabolism and clearance. The reaction must be fast, on the time scale of cellular processes (minutes) to prevent competition in reactions which may diminish the small signals of less abundant species. Rapid reactions also offer a fast response, necessary in order to accurately track dynamic processes.
Reaction biocompatibility: Reactions have to be non-toxic and must function in biological conditions taking into account pH, aqueous environments, and temperature. Pharmacokinetics are a growing concern as bioorthogonal chemistry expands to live animal models.
Accessible engineering: The chemical reporter must be capable of incorporation into biomolecules via some form of metabolic or protein engineering. Optimally, one of the functional groups is also very small so that it does not disturb native behavior.
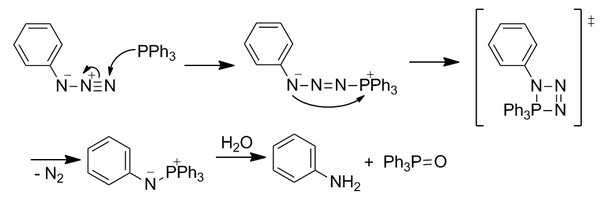
Staudinger ligation
The Staudinger ligation is a reaction developed by the Bertozzi group in 2000 that is based on the classic Staudinger reaction of azides with triarylphosphines.[15] It launched the field of bioorthogonal chemistry as the first reaction with completely abiotic functional groups although it is no longer as widely used. The Staudinger ligation has been used in both live cells and live mice.[5]
Bioorthogonality
The azide can act as a soft electrophile that prefers soft nucleophiles such as phosphines. This is in contrast to most biological nucleophiles which are typically hard nucleophiles. The reaction proceeds selectively under water-tolerant conditions to produce a stable product.
Phosphines are completely absent from living systems and do not reduce disulfide bonds despite mild reduction potential. Azides had been shown to be biocompatible in FDA-approved drugs such as azidothymidine and through other uses as cross linkers. Additionally, their small size allows them to be easily incorporated into biomolecules through cellular metabolic pathways.
The nucleophilic phosphine attacks the azide at the electrophilic terminal nitrogen. Through a four-membered transition state, N2 is lost to form an aza-ylide. The unstable ylide is hydrolyzed to form phosphine oxide and a primary amine. However, this reaction is not immediately bioorthogonal because hydrolysis breaks the covalent bond in the aza-ylide.
Staudinger ligation
The reaction was modified to include an ester group ortho to the phosphorus atom on one of the aryl rings to direct the aza-ylide through a new path of reactivity in order to outcompete immediate hydrolysis by positioning the ester to increase local concentration. The initial nucleophilic attack on the azide is the rate-limiting step. The ylide reacts with the electrophilic ester trap through intramolecular cyclization to form a five-membered ring. This ring undergoes hydrolysis to form a stable amide bond.
Limitations
The phosphine reagents slowly undergo air oxidation in living systems. Additionally, it is likely that they are metabolized in vitro by cytochrome P450 enzymes.
The kinetics of the reactions are slow with second order rate constants around 0.0020M−1•s−1. Attempts to increase nucleophilic attack rates by adding electron-donating groups to the phosphines improved kinetics, but also increased the rate of air oxidation.
The poor kinetics require that high concentrations of the phosphine be used which leads to problems with high background signal in imaging applications. Attempts have been made to combat the problem of high background through the development of a fluorogenic phosphine reagents based on fluorescein and luciferin, but the intrinsic kinetics remain a limitation.[16]
Copper-free click chemistry is a bioorthogonal reaction first developed by Carolyn Bertozzi as an activated variant of an azide alkyne Huisgen cycloaddition, based on the work by Karl Barry Sharpless et al. Unlike CuAAC, Cu-free click chemistry has been modified to be bioorthogonal by eliminating a cytotoxic copper catalyst, allowing reaction to proceed quickly and without live cell toxicity. Instead of copper, the reaction is a strain-promoted alkyne-azide cycloaddition (SPAAC). It was developed as a faster alternative to the Staudinger ligation, with the first generations reacting over sixty times faster. The bioorthogonality of the reaction has allowed the Cu-free click reaction to be applied within cultured cells, live zebrafish, and mice.
click chemistry labeling
Copper toxicity
The classic copper-catalyzed azide-alkyne cycloaddition has been an extremely fast and effective click reaction for bioconjugation, but it is not suitable for use in live cells due to the toxicity of Cu(I) ions. Toxicity is due to oxidative damage from reactive oxygen species formed by the copper catalysts. Copper complexes have also been found to induce changes in cellular metabolism and are taken up by cells.
There has been some development of ligands to prevent biomolecule damage and facilitate removal in in vitro applications. However, it has been found that different ligand environments of complexes can still affect metabolism and uptake, introducing an unwelcome perturbation in cellular function.[17]
Bioorthogonality
The azide group is particularly bioorthogonal because it is extremely small (favorable for cell permeability and avoids perturbations), metabolically stable, and does not naturally exist in cells and thus has no competing biological side reactions. Although azides are not the most reactive 1,3-dipole available for reaction, they are preferred for their relative lack of side reactions and stability in typical synthetic conditions.[18] The alkyne is not as small, but it still has the stability and orthogonality necessary for in vivo labeling. Cyclooctynes are traditionally the most common cycloalkyne for labeling studies, as they are the smallest stable alkyne ring.
Mechanism
The reaction proceeds as a standard 1,3-dipolar cycloaddition, a type of asynchronous, concerted pericyclic shift. The ambivalent nature of the 1,3-dipole should make the identification of an electrophilic or nucleophilic center on the azide impossible such that the direction of the cyclic electron flow is meaningless. [p] However, computation has shown that the electron distribution amongst nitrogens causes the innermost nitrogen atom to bear the greatest negative charge.[19]
Regioselectivity
Although the reaction produces a regioisomeric mixture of triazoles, the lack of regioselectivity in the reaction is not a major concern for most current applications. More regiospecific and less bioorthogonal requirements are best served by copper-catalyzed Huisgen cycloaddition, especially given the synthetic difficulty (compared to the addition of a terminal alkyne) of synthesizing a strained cyclooctyne.
Development of cyclooctynes
Cyclooctyne
Second order rate constant (M−1s−1)
OCT
0.0024
ALO
0.0013
MOFO
0.0043
DIFO
0.076
DIBO
0.057
BARAC
0.96
DIBAC (ADIBO)
0.31
DIMAC
0.0030
Strained cyclooctynes developed for copper-free click chemistry
OCT was the first cyclooctyne developed for Cu-free click chemistry. While linear alkynes are unreactive at physiological temperatures, OCT was able readily react with azides in biological conditions while showing no toxicity. However, it was poorly water-soluble, and the kinetics were barely improved over the Staudinger ligation. ALO (aryl-less octyne) was developed to improve water solubility, but it still had poor kinetics.
Monofluorinated (MOFO) and difluorinated (DIFO) cyclooctynes were created to increase the rate through the addition of electron-withdrawing fluorine substituents at the propargylic position. Fluorine is a good electron-withdrawing group in terms of synthetic accessibility and biological inertness. In particular, it cannot form an electrophilic Michael acceptor that may side-react with biological nucleophiles.[8]DIBO (dibenzocyclooctyne) was developed as a fusion to two aryl rings, resulting in very high strain and a decrease in distortion energies. It was proposed that biaryl substitution increases ring strain and provides conjugation with the alkyne to improve reactivity. Although calculations have predicted that mono-aryl substitution would provide an optimal balance between steric clash (with azide molecule) and strain,[20] monoarylated products have been shown to be unstable.
BARAC (biarylazacyclooctynone) followed with the addition of an amide bond which adds an sp2-like center to increase rate by distortion. Amide resonance contributes additional strain without creating additional unsaturation which would lead to an unstable molecule. Additionally, the addition of a heteroatom into the cyclooctyne ring improves both solubility and pharmacokinetics of the molecule. BARAC has sufficient rate (and sensitivity) to the extent that washing away excess probe is unnecessary to reduce background. This makes it extremely useful in situations where washing is impossible as in real-time imaging or whole animal imaging. Although BARAC is extremely useful, its low stability requires that it must be stored at 0°C, protected from light and oxygen.[21]
The synthesis was designed by the Bertozzi group as a modular route to facilitate future modifications in SAR analysis. The first step is Fischer indole synthesis. The product is alkylated with allyl bromide as a handle for future probe attachment; TMS is then added. Oxidation opens the central rings to form a cyclic amide. The ketone is treated as an enolate to add a triflate group. Reaction of the terminal alkene generates a linker for conjugation to a molecule. The final reaction with CsF introduces the strained alkyne at the last step.
Further adjustments variations on BARAC to produce DIBAC/ADIBO were performed to add distal ring strain and reduce sterics around the alkyne to further increase reactivity. Keto-DIBO, in which the hydroxyl group has been converted to a ketone, has a three-fold increase in rate due to a change in ring conformation. Attempts to make a difluorobenzocyclooctyne (DIFBO) were unsuccessful due to the instability.
Problems with DIFO with in vivo mouse studies illustrate the difficulty of producing bioorthogonal reactions. Although DIFO was extremely reactive in the labeling of cells, it performed poorly in mouse studies due to binding with serum albumin. Hydrophobicity of the cyclooctyne promotes sequestration by membranes and serum proteins, reducing bioavailable concentrations. In response, DIMAC (dimethoxyazacyclooctyne) was developed to increase water solubility, polarity, and pharmacokinetics,[22] although efforts in bioorthogonal labeling of mouse models is still in development.
Reactivity
Computational efforts have been vital in explaining the thermodynamics and kinetics of these cycloaddition reactions which has played a vital role in continuing to improve the reaction. There are two methods for activating alkynes without sacrificing stability: decrease transition state energy or decrease reactant stability.
The red arrow shows the direction of energy change. Black arrows show the difference in activation energy before and after the effects.
Decreasing reactant stability:Houk[23] has proposed that differences in the energy (Ed ‡) required to distort the azide and alkyne into the transition state geometries control the barrier heights for the reaction. The activation energy (E ‡) is the sum of destabilizing distortions and stabilizing interactions (Ei ‡). The most significant distortion is in the azide functional group with lesser contribution of alkyne distortion. However, it is only the cyclooctyne that can be easily modified for higher reactivity. Calculated barriers of reaction for phenyl azide and acetylene (16.2 kcal/mol) versus cyclooctyne (8.0 kcal/mol) results in a predicted rate increase of 106. The cyclooctyne requires less distortion energy (1.4 kcal/mol versus 4.6 kcal/mol) resulting in a lower activation energy despite smaller interaction energy.
Relationship between activation energy, distortion energy, and interaction energy
Decreasing transition state energy: Electron withdrawing groups such as fluorine increase rate by decreasing LUMO energy and the HOMO-LUMO gap. This leads to a greater charge transfer from the azide to the fluorinated cyclooctyne in the transition state, increasing interaction energy (lower negative value) and overall activation energy.[24] The lowering of the LUMO is the result of hyperconjugation between alkyne π donor orbitals and CF σ* acceptors. These interactions provide stabilization primarily in the transition state as a result of increased donor/acceptor abilities of the bonds as they distort. NBO calculations have shown that transition state distortion increases the interaction energy by 2.8 kcal/mol.
The hyperconjugation between out-of-plane π bonds is greater because the in-plane π bonds are poorly aligned. However, transition state bending allows the in-plane π bonds to have a more antiperiplanar arrangement that facilitates interaction. Additional hyperconjugative interaction energy stabilization is achieved through an increase in the electronic population of the σ* due to the forming CN bond. Negative hyperconjugation with the σ* CF bonds enhances this stabilizing interaction.[19]
Regioselectivity
Although regioselectivity is not a great issue in the current imaging applications of copper-free click chemistry, it is an issue that prevents future applications in fields such as drug design or peptidomimetics.[25]
Currently most cyclooctynes react to form regioisomeric mixtures. [m] Computation analysis has found that while gas phase regioselectivity is calculated to favor 1,5 addition over 1,4 addition by up to 2.9 kcal/mol in activation energy, solvation corrections result in the same energy barriers for both regioisomers. While the 1,4 isomer in the cycloaddition of DIFO is disfavored by its larger dipole moment, solvation stabilizes it more strongly than the 1,5 isomer, eroding regioselectivity.[24]
Symmetrical cyclooctynes such as BCN (bicyclo[6.1.0]nonyne) form a single regioisomer upon cycloaddition[26] and may serve to address this problem in the future.
Applications
The most widespread application of copper-free click chemistry is in biological imaging in live cells or animals using an azide-tagged biomolecule and a cyclooctyne bearing an imaging agent.
Fluorescent keto and oxime variants of DIBO are used in fluoro-switch click reactions in which the fluorescence of the cyclooctyne is quenched by the triazole that forms in the reaction.[27] On the other hand, coumarin-conjugated cyclooctynes such as coumBARAC have been developed such that the alkyne suppresses fluorescence while triazole formation increases the fluorescence quantum yield by ten-fold.[28]
coumBARAC fluorescence increases with reaction
Spatial and temporal control of substrate labeling has been investigated using photoactivatable cyclooctynes. This allows equilibration of the alkyne prior to reaction in order to reduce artifacts as a result of concentration gradients. Masked cyclooctynes are unable to react with azides in the dark but become reactive alkynes upon irradiation with light.[29]
Copper-free click chemistry is being explored for use in synthesizing PET imaging agents which must be made quickly with high purity and yield in order to minimize isotopic decay before the compounds can be administered. Both the high rate constants and the bioorthogonality of SPAAC are amenable to PET chemistry.[30]
Other bioorthogonal reactions
Nitrone dipole cycloaddition
Copper-free click chemistry has been adapted to use nitrones as the 1,3-dipole rather than azides and has been used in the modification of peptides.[9]
This cycloaddition between a nitrone and a cyclooctyne forms N-alkylated isoxazolines. The reaction rate is enhanced by water and is extremely fast with second order rate constants ranging from 12 to 32M−1•s−1, depending on the substitution of the nitrone. Although the reaction is extremely fast, it faces problems in incorporating the nitrone into biomolecules through metabolic labeling. Labeling has only been achieved through post-translational peptide modification.
Norbornene cycloaddition
1,3 dipolar cycloadditions have been developed as a bioorthogonal reaction using a nitrile oxide as a 1,3-dipole and a norbornene as a dipolarophile. Its primary use has been in labeling DNA and RNA in automated oligonucleotide synthesizers,[31] and polymer crosslinking in the presence of living cells.[32]
Norbornenes were selected as dipolarophiles due to their balance between strain-promoted reactivity and stability. The drawbacks of this reaction include the cross-reactivity of the nitrile oxide due to strong electrophilicity and slow reaction kinetics.
Oxanorbornadiene cycloaddition
The oxanorbornadiene cycloaddition is a 1,3-dipolar cycloaddition followed by a retro-Diels Alder reaction to generate a triazole-linked conjugate with the elimination of a furan molecule.[33] Preliminary work has established its usefulness in peptide labeling experiments, and it has also been used in the generation of SPECT imaging compounds.[34] More recently, the use of an oxanorbornadiene was described in a catalyst-free room temperature "iClick" reaction, in which a model amino acid is linked to the metal moiety, in a novel approach to bioorthogonal reactions.[35]
Ring strain and electron deficiency in the oxanorbornadiene increase reactivity towards the cycloaddition rate-limiting step. The retro-Diels Alder reaction occurs quickly afterwards to form the stable 1,2,3 triazole. Problems include poor tolerance for substituents which may change electronics of the oxanorbornadiene and low rates (second order rate constants on the order of 10−4).
Tetrazine ligation
The tetrazine ligation is the reaction of a trans-cyclooctene and an s-tetrazine in an inverse-demand Diels Alder reaction followed by a retro-Diels Alder reaction to eliminate nitrogen gas.[36] The reaction is extremely rapid with a second order rate constant of 2000M−1–s−1 (in 9:1 methanol/water) allowing modifications of biomolecules at extremely low concentrations.
Based on computational work by Bach, the strain energy for Z-cyclooctenes is 7.0 kcal/mol compared to 12.4 kcal/mol for cyclooctane due to a loss of two transannular interactions. E-cyclooctene has a highly twisted double bond resulting in a strain energy of 17.9 kcal/mol.[37] As such, the highly strained trans-cyclooctene is used as a reactive dienophile. The diene is a 3,6-diaryl-s-tetrazine which has been substituted in order to resist immediate reaction with water. The reaction proceeds through an initial cycloaddition followed by a reverse Diels Alder to eliminate N2 and prevent reversibility of the reaction.[11]
Not only is the reaction tolerant of water, but it has been found that the rate increases in aqueous media. Reactions have also been performed using norbornenes as dienophiles at second order rates on the order of 1M−1•s−1 in aqueous media. The reaction has been applied in labeling live cells[38] and polymer coupling.[39]
[4+1] Cycloaddition
This isocyanide click reaction is a [4+1] cycloaddition followed by a retro-Diels Alder elimination of N2.[12]
The reaction proceeds with an initial [4+1] cycloaddition followed by a reversion to eliminate a thermodynamic sink and prevent reversibility. This product is stable if a tertiary amine or isocyanopropanoate is used. If a secondary or primary isocyanide is used, the produce will form an imine which is quickly hydrolyzed.
Isocyanide is a favored chemical reporter due to its small size, stability, non-toxicity, and absence in mammalian systems. However, the reaction is slow, with second order rate constants on the order of 10−2M−1•s−1.
Tetrazole photoclick chemistry
Photoclick chemistry utilizes a photoinduced cycloelimination to release N2. This generates a short-lived 1,3 nitrile imine intermediate via the loss of nitrogen gas, which undergoes a 1,3-dipolar cycloaddition with an alkene to generate pyrazoline cycloadducts.[12]
Photoinduction takes place with a brief exposure to light (wavelength is tetrazole-dependent) to minimize photodamage to cells. The reaction is enhanced in aqueous conditions and generates a single regioisomer.
The transient nitrile imine is highly reactive for 1,3-dipolar cycloaddition due to a bent structure which reduces distortion energy. Substitution with electron-donating groups on phenyl rings increases the HOMO energy, when placed on the 1,3 nitrile imine and increases the rate of reaction.
Advantages of this approach include the ability to spatially or temporally control reaction and the ability to incorporate both alkenes and tetrazoles into biomolecules using simple biological methods such as genetic encoding.[40] Additionally, the tetrazole can be designed to be fluorogenic in order to monitor progress of the reaction.[41]
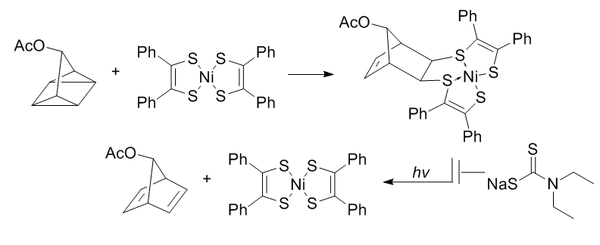
Quadricyclane ligation
The quadricyclane ligation utilizes a highly strained quadricyclane to undergo [2+2+2] cycloaddition with π systems.[13]
Quadricyclane is abiotic, unreactive with biomolecules (due to complete saturation), relatively small, and highly strained (~80 kcal/mol). However, it is highly stable at room temperature and in aqueous conditions at physiological pH. It is selectively able to react with electron-poor π systems but not simple alkenes, alkynes, or cyclooctynes.
Bis(dithiobenzil)nickel(II) was chosen as a reaction partner out of a candidate screen based on reactivity. To prevent light-induced reversion to norbornadiene, diethyldithiocarbamate is added to chelate the nickel in the product.
These reactions are enhanced by aqueous conditions with a second order rate constant of 0.25M−1•s−1. Of particular interest is that it has been proven to be bioorthogonal to both oxime formation and copper-free click chemistry.
↑ Kennedy, David C.; McKay, Craig S.; Legault, Marc C. B.; Danielson, Dana C.; Blake, Jessie A.; Pegoraro, Adrian F.; Stolow, Albert; Mester, Zoltan; Pezacki, John Paul (2011). "Cellular Consequences of Copper Complexes Used to Catalyze Bioorthogonal Click Reactions". Journal of the American Chemical Society. 133 (44): 17993–8001. Bibcode:2011JAChS.13317993K. doi:10.1021/ja2083027. PMID21970470.
↑ Huisgen, Rolf. (1976). "1,3-Dipolar cycloadditions. 76. Concerted nature of 1,3-dipolar cycloadditions and the question of diradical intermediates". The Journal of Organic Chemistry. 41 (3): 403–419. doi:10.1021/jo00865a001.
1 2 Gold, Brian; Shevchenko, Nikolay E.; Bonus, Natalie; Dudley, Gregory B.; Alabugin, Igor V. (2011). "Selective Transition State Stabilization via Hyperconjugative and Conjugative Assistance: Stereoelectronic Concept for Copper-Free Click Chemistry". The Journal of Organic Chemistry. 77 (1): 75–89. doi:10.1021/jo201434w. PMID22077877.
↑ Ess, Daniel H.; Jones, Gavin O.; Houk, K. N. (2008). "Transition States of Strain-Promoted Metal-Free Click Chemistry: 1,3-Dipolar Cycloadditions of Phenyl Azide and Cyclooctynes". Organic Letters. 10 (8): 1633–6. doi:10.1021/ol8003657. PMID18363405.
1 2 Schoenebeck, Franziska; Ess, Daniel H.; Jones, Gavin O.; Houk, K. N. (2009). "Reactivity and Regioselectivity in 1,3-Dipolar Cycloadditions of Azides to Strained Alkynes and Alkenes: A Computational Study". Journal of the American Chemical Society. 131 (23): 8121–33. Bibcode:2009JAChS.131.8121S. doi:10.1021/ja9003624. PMID19459632.
↑ Gutsmiedl, Katrin; Wirges, Christian T.; Ehmke, Veronika; Carell, Thomas (2009). "Copper-Free "Click" Modification of DNA via Nitrile Oxide Norbornene 1,3-Dipolar Cycloaddition". Organic Letters. 11 (11): 2405–8. doi:10.1021/ol9005322. PMID19405510.
↑ Truong, Vinh X.; Zhou, Kun; Simon P., George; Forsythe, John S. (2015). "Nitrile Oxide-Norbornene Cycloaddition as a Bioorthogonal Crosslinking Reaction for the Preparation of Hydrogels". Macromolecular Rapid Communications. 36 (19): 1729–34. doi:10.1002/marc.201500314. PMID26250120.
↑ Van Berkel, Sander S.; Dirks, A. (Ton) J.; Meeuwissen, Silvie A.; Pingen, Dennis L. L.; Boerman, Otto C.; Laverman, Peter; Van Delft, Floris L.; Cornelissen, Jeroen J. L. M.; Rutjes, Floris P. J. T. (2008). "Application of Metal Free Triazole Formation in the Synthesis of Cyclic RGD DTPA Conjugates". ChemBioChem. 9 (11): 1805–15. doi:10.1002/cbic.200800074. hdl:2066/69881. PMID18623291. S2CID205552916.
↑ Bach, Robert D. (2009). "Ring Strain Energy in the Cyclooctyl System. The Effect of Strain Energy on [3 + 2] Cycloaddition Reactions with Azides". Journal of the American Chemical Society. 131 (14): 5233–43. Bibcode:2009JAChS.131.5233B. doi:10.1021/ja8094137. PMID19301865.
↑ Hansell, Claire F.; Espeel, Pieter; Stamenovic, Milan M.; Barker, Ian A.; Dove, Andrew P.; Du Prez, Filip E.; o Reilly, Rachel K. (2011). "Additive-Free Clicking for Polymer Functionalization and Coupling by Tetrazine Norbornene Chemistry". Journal of the American Chemical Society. 133 (35): 13828–31. Bibcode:2011JAChS.13313828H. doi:10.1021/ja203957h. PMID21819063.
↑ Song, Wenjiao; Wang, Yizhong; Qu, Jun; Lin, Qing (2008). "Selective Functionalization of a Genetically Encoded Alkene-Containing Protein via "Photoclick Chemistry" in Bacterial Cells". Journal of the American Chemical Society. 130 (30): 9654–5. Bibcode:2008JAChS.130.9654S. doi:10.1021/ja803598e. PMID18593155.
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.