is the pressure of the system (which must be equal to that of the surroundings for mechanical equilibrium).
The Gibbs free energy change (, measured in joules in SI) is the maximum amount of non-volume expansion work that can be extracted from a closed system (one that can exchange heat and work with its surroundings, but not matter) at fixed temperature and pressure. This maximum can be attained only in a completely reversible process. When a system transforms reversibly from an initial state to a final state under these conditions, the decrease in Gibbs free energy equals the work done by the system to its surroundings, minus the work of the pressure forces.[1]
The Gibbs energy is the thermodynamic potential that is minimized when a system reaches chemical equilibrium at constant pressure and temperature when not driven by an applied electrolytic voltage. Its derivative with respect to the reaction coordinate of the system then vanishes at the equilibrium point. As such, a reduction in is necessary for a reaction to be spontaneous under these conditions.
The concept of Gibbs free energy, originally called available energy, was developed in the 1870s by the American scientist Josiah Willard Gibbs. In 1873, Gibbs described this "available energy" as[2]:400
the greatest amount of mechanical work which can be obtained from a given quantity of a certain substance in a given initial state, without increasing its total volume or allowing heat to pass to or from external bodies, except such as at the close of the processes are left in their initial condition.
The reaction C(s)→C(s) has a negative change in Gibbs free energy and is therefore thermodynamically favorable at 25°C and 1 atm. However, the reaction is too slow to be observed, because of its very high activation energy. Whether a reaction is thermodynamically favorable does not determine its rate.
According to the second law of thermodynamics, for systems reacting at fixed temperature and pressure without input of non-Pressure Volume (pV) work, there is a general natural tendency to achieve a minimum of the Gibbs free energy.
A quantitative measure of the favorability of a given reaction under these conditions is the change ΔG (sometimes written "delta G" or "dG") in Gibbs free energy that is (or would be) caused by the reaction. As a necessary condition for the reaction to occur at constant temperature and pressure, ΔG must be smaller than the non-pressure-volume (non-pV, e.g. electrical) work, which is often equal to zero (then ΔG must be negative). ΔG equals the maximum amount of non-pV work that can be performed as a result of the chemical reaction for the case of a reversible process. If analysis indicates a positive ΔG for a reaction, then energy — in the form of electrical or other non-pV work — would have to be added to the reacting system for ΔG to be smaller than the non-pV work and make it possible for the reaction to occur.[3]:298–299
One can think of ∆G as the amount of "free" or "useful" energy available to do non-pV work at constant temperature and pressure. The equation can be also seen from the perspective of the system taken together with its surroundings (the rest of the universe). First, one assumes that the given reaction at constant temperature and pressure is the only one that is occurring. Then the entropy released or absorbed by the system equals the entropy that the environment must absorb or release, respectively. The reaction will only be allowed if the total entropy change of the universe is zero or positive. This is reflected in a negative ΔG, and the reaction is called an exergonic process.
If two chemical reactions are coupled, then an otherwise endergonic reaction (one with positive ΔG) can be made to happen. The input of heat into an inherently endergonic reaction, such as the elimination of cyclohexanol to cyclohexene, can be seen as coupling an unfavorable reaction (elimination) to a favorable one (burning of coal or other provision of heat) such that the total entropy change of the universe is greater than or equal to zero, making the total Gibbs free energy change of the coupled reactions negative.
In traditional use, the term "free" was included in "Gibbs free energy" to mean "available in the form of useful work".[1] The characterization becomes more precise if we add the qualification that it is the energy available for non-pressure-volume work.[4] (An analogous, but slightly different, meaning of "free" applies in conjunction with the Helmholtz free energy, for systems at constant temperature). However, an increasing number of books and journal articles do not include the attachment "free", referring to G as simply "Gibbs energy". This is the result of a 1988 IUPAC meeting to set unified terminologies for the international scientific community, in which the removal of the adjective "free" was recommended.[5][6][7] This standard, however, has not yet been universally adopted.
The name "free enthalpy" was also used for G in the past.[6]
The quantity called "free energy" is a more advanced and accurate replacement for the outdated term affinity, which was used by chemists in the earlier years of physical chemistry to describe the force that caused chemical reactions.
In 1873, Josiah Willard Gibbs published A Method of Geometrical Representation of the Thermodynamic Properties of Substances by Means of Surfaces, in which he sketched the principles of his new equation that was able to predict or estimate the tendencies of various natural processes to ensue when bodies or systems are brought into contact. By studying the interactions of homogeneous substances in contact, i.e., bodies composed of part solid, part liquid, and part vapor, and by using a three-dimensional volume-entropy-internal energy graph, Gibbs was able to determine three states of equilibrium, i.e., "necessarily stable", "neutral", and "unstable", and whether or not changes would ensue. Further, Gibbs stated:[2]
If we wish to express in a single equation the necessary and sufficient condition of thermodynamic equilibrium for a substance when surrounded by a medium of constant pressurep and temperatureT, this equation may be written:
δ(ε − Tη + pν) = 0
when δ refers to the variation produced by any variations in the state of the parts of the body, and (when different parts of the body are in different states) in the proportion in which the body is divided between the different states. The condition of stable equilibrium is that the value of the expression in the parenthesis shall be a minimum.
In this description, as used by Gibbs, ε refers to the internal energy of the body, η refers to the entropy of the body, and ν is the volume of the body...
Thereafter, in 1882, the German scientist Hermann von Helmholtz characterized the affinity as the largest quantity of work which can be gained when the reaction is carried out in a reversible manner, e.g., electrical work in a reversible cell. The maximum work is thus regarded as the diminution of the free, or available, energy of the system (Gibbs free energyG at T = constant, P = constant or Helmholtz free energyF at T = constant, V = constant), whilst the heat given out is usually a measure of the diminution of the total energy of the system (internal energy). Thus, G or F is the amount of energy "free" for work under the given conditions.
Until this point, the general view had been such that: "all chemical reactions drive the system to a state of equilibrium in which the affinities of the reactions vanish". Over the next 60 years, the term affinity came to be replaced with the term free energy. According to chemistry historian Henry Leicester, the influential 1923 textbook Thermodynamics and the Free Energy of Chemical Substances by Gilbert N. Lewis and Merle Randall led to the replacement of the term "affinity" by the term "free energy" in much of the English-speaking world.[8]:206
Definitions
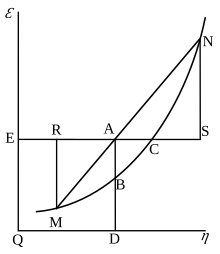
Willard Gibbs' 1873 available energy (free energy) graph, which shows a plane perpendicular to the axis of v (volume) and passing through point A, which represents the initial state of the body. MN is the section of the surface of dissipated energy. Qε and Qη are sections of the planes η = 0 and ε = 0, and therefore parallel to the axes of ε (internal energy) and η (entropy), respectively. AD and AE are the energy and entropy of the body in its initial state, AB and AC its available energy (Gibbs free energy) and its capacity for entropy (the amount by which the entropy of the body can be increased without changing the energy of the body or increasing its volume), respectively.
The expression for the infinitesimal reversible change in the Gibbs free energy as a function of its "natural variables"p and T, for an open system, subjected to the operation of external forces (for instance, electrical or magnetic) Xi, which cause the external parameters of the system ai to change by an amount dai, can be derived as follows from the first law for reversible processes:
Ni is the number of particles (or number of moles) composing the i-th chemical component.
This is one form of the Gibbs fundamental equation.[10] In the infinitesimal expression, the term involving the chemical potential accounts for changes in Gibbs free energy resulting from an influx or outflux of particles. In other words, it holds for an open system or for a closed, chemically reacting system where the Ni are changing. For a closed, non-reacting system, this term may be dropped.
Any number of extra terms may be added, depending on the particular system being considered. Aside from mechanical work, a system may, in addition, perform numerous other types of work. For example, in the infinitesimal expression, the contractile work energy associated with a thermodynamic system that is a contractile fiber that shortens by an amount −dl under a force f would result in a term f dl being added. If a quantity of charge −de is acquired by a system at an electrical potential Ψ, the electrical work associated with this is −Ψ de, which would be included in the infinitesimal expression. Other work terms are added on per system requirements.[11]
Each quantity in the equations above can be divided by the amount of substance, measured in moles, to form molar Gibbs free energy. The Gibbs free energy is one of the most important thermodynamic functions for the characterization of a system. It is a factor in determining outcomes such as the voltage of an electrochemical cell, and the equilibrium constant for a reversible reaction. In isothermal, isobaric systems, Gibbs free energy can be thought of as a "dynamic" quantity, in that it is a representative measure of the competing effects of the enthalpic[clarification needed] and entropic driving forces involved in a thermodynamic process.
Relation to other relevant parameters
The temperature dependence of the Gibbs energy for an ideal gas is given by the Gibbs–Helmholtz equation, and its pressure dependence is given by[12]
Because some of the natural variables of G are intensive, dG may not be integrated using Euler relations as is the case with internal energy. However, simply substituting the above integrated result for U into the definition of G gives a standard expression for G:[13]
This result shows that the chemical potential of a substance is its (partial) mol(ecul)ar Gibbs free energy. It applies to homogeneous, macroscopic systems, but not to all thermodynamic systems.[14]
Gibbs free energy of reactions
The system under consideration is held at constant temperature and pressure, and is closed (no matter can come in or out). The Gibbs energy of any system is and an infinitesimal change in G, at constant temperature and pressure, yields
where δQ is energy added as heat, and δW is energy added as work. The work done on the system may be written as δW = −pdV + δWx, where −pdV is the mechanical work of compression/expansion done on or by the system and δWx is all other forms of work, which may include electrical, magnetic, etc. Then
and the infinitesimal change in G is
.
The second law of thermodynamics states that for a closed system at constant temperature (in a heat bath), , and so it follows that
Assuming that only mechanical work is done, this simplifies to
This means that for such a system when not in equilibrium, the Gibbs energy will always be decreasing, and in equilibrium, the infinitesimal change dG will be zero. In particular, this will be true if the system is experiencing any number of internal chemical reactions on its path to equilibrium.
In electrochemical thermodynamics
When electric charge dQele is passed between the electrodes of an electrochemical cell generating an emf, an electrical work term appears in the expression for the change in Gibbs energy: where S is the entropy, V is the system volume, p is its pressure and T is its absolute temperature.
The combination (, Qele) is an example of a conjugate pair of variables. At constant pressure the above equation produces a Maxwell relation that links the change in open cell voltage with temperature T (a measurable quantity) to the change in entropy S when charge is passed isothermally and isobarically. The latter is closely related to the reaction entropy of the electrochemical reaction that lends the battery its power. This Maxwell relation is:[15]
If a mole of ions goes into solution (for example, in a Daniell cell, as discussed below) the charge through the external circuit is
where n0 is the number of electrons/ion, and F0 is the Faraday constant and the minus sign indicates discharge of the cell. Assuming constant pressure and volume, the thermodynamic properties of the cell are related strictly to the behavior of its emf by
where ΔH is the enthalpy of reaction. The quantities on the right are all directly measurable.
Useful identities to derive the Nernst equation
This section may be confusing or unclear to readers. In particular, the physical situation is not explained. Also, the circle notation is not well explained (even in the one case where it is attempted). It's just bare equations. Please help clarify the section. There might be a discussion about this on the talk page.(March 2015) ( Learn how and when to remove this message )
During a reversible electrochemical reaction at constant temperature and pressure, the following equations involving the Gibbs free energy hold:
(for a reversible electrochemical process at constant temperature and pressure),
(definition of ),
and rearranging gives
which relates the cell potential resulting from the reaction to the equilibrium constant and reaction quotient for that reaction (Nernst equation),
where
ΔrG, Gibbs free energy change per mole of reaction,
ΔrG°, Gibbs free energy change per mole of reaction for unmixed reactants and products at standard conditions (i.e. 298K, 100kPa, 1M of each reactant and product),
The standard Gibbs free energy of formation of a compound is the change of Gibbs free energy that accompanies the formation of 1 mole of that substance from its component elements, in their standard states (the most stable form of the element at 25°C and 100kPa). Its symbol is ΔfG˚.
All elements in their standard states (diatomic oxygen gas, graphite, etc.) have standard Gibbs free energy change of formation equal to zero, as there is no change involved.
At equilibrium, ΔfG = 0, and Qf = K, so the equation becomes
ΔfG˚ = −RT ln K,
where K is the equilibrium constant of the formation reaction of the substance from the elements in their standard states.
Graphical interpretation by Gibbs
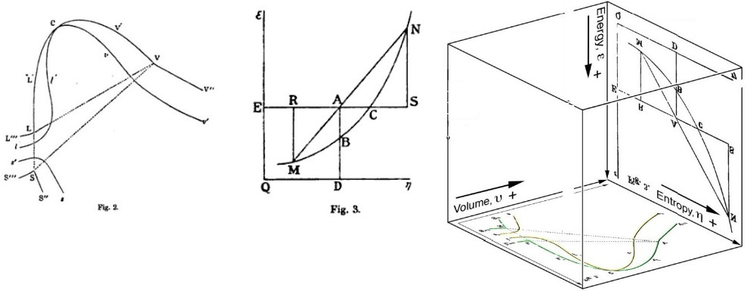
Gibbs free energy was originally defined graphically. In 1873, American scientist Willard Gibbs published his first thermodynamics paper, "Graphical Methods in the Thermodynamics of Fluids", in which Gibbs used the two coordinates of the entropy and volume to represent the state of the body. In his second follow-up paper, "A Method of Geometrical Representation of the Thermodynamic Properties of Substances by Means of Surfaces", published later that year, Gibbs added in the third coordinate of the energy of the body, defined on three figures. In 1874, Scottish physicist James Clerk Maxwell used Gibbs' figures to make a 3D energy-entropy-volume thermodynamic surface of a fictitious water-like substance.[17] Thus, in order to understand the concept of Gibbs free energy, it may help to understand its interpretation by Gibbs as section AB on his figure 3, and as Maxwell sculpted that section on his 3D surface figure.
American scientist Willard Gibbs' 1873 figures two and three (above left and middle) used by Scottish physicist James Clerk Maxwell in 1874 to create a three-dimensional entropy, volume, energythermodynamic surface diagram for a fictitious water-like substance, transposed the two figures of Gibbs (above right) onto the volume-entropy coordinates (transposed to bottom of cube) and energy-entropy coordinates (flipped upside down and transposed to back of cube), respectively, of a three-dimensional Cartesian coordinates; the region AB being the first-ever three-dimensional representation of Gibbs free energy, or what Gibbs called "available energy"; the region AC being its capacity for entropy, what Gibbs defined as "the amount by which the entropy of the body can be increased without changing the energy of the body or increasing its volume.
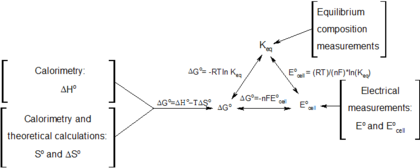
1 2 "Gibbs energy (function), G". IUPAC Gold Book (Compendium of Chemical Technology). IUPAC (International Union of Pure and Applied Chemistry). 2008. doi:10.1351/goldbook.G02629. Retrieved 24 December 2020. It was formerly called free energy or free enthalpy.
In a chemical reaction, chemical equilibrium is the state in which both the reactants and products are present in concentrations which have no further tendency to change with time, so that there is no observable change in the properties of the system. This state results when the forward reaction proceeds at the same rate as the reverse reaction. The reaction rates of the forward and backward reactions are generally not zero, but they are equal. Thus, there are no net changes in the concentrations of the reactants and products. Such a state is known as dynamic equilibrium.
Chemical thermodynamics is the study of the interrelation of heat and work with chemical reactions or with physical changes of state within the confines of the laws of thermodynamics. Chemical thermodynamics involves not only laboratory measurements of various thermodynamic properties, but also the application of mathematical methods to the study of chemical questions and the spontaneity of processes.
Enthalpy is the sum of a thermodynamic system's internal energy and the product of its pressure and volume. It is a state function in thermodynamics used in many measurements in chemical, biological, and physical systems at a constant external pressure, which is conveniently provided by the large ambient atmosphere. The pressure–volume term expresses the work that was done against constant external pressure to establish the system's physical dimensions from to some final volume , i.e. to make room for it by displacing its surroundings. The pressure-volume term is very small for solids and liquids at common conditions, and fairly small for gases. Therefore, enthalpy is a stand-in for energy in chemical systems; bond, lattice, solvation, and other chemical "energies" are actually enthalpy differences. As a state function, enthalpy depends only on the final configuration of internal energy, pressure, and volume, not on the path taken to achieve it.
Le Chatelier's principle, also called Chatelier's principle, is a principle of chemistry used to predict the effect of a change in conditions on chemical equilibrium. The principle is named after French chemist Henry Louis Le Chatelier, and sometimes also credited to Karl Ferdinand Braun, who discovered it independently. It can be defined as:
If the equilibrium of a system is disturbed by a change in one or more of the determining factors the system tends to adjust itself to a new equilibrium by counteracting as far as possible the effect of the change
In thermodynamics, the thermodynamic free energy is one of the state functions of a thermodynamic system. The change in the free energy is the maximum amount of work that the system can perform in a process at constant temperature, and its sign indicates whether the process is thermodynamically favorable or forbidden. Since free energy usually contains potential energy, it is not absolute but depends on the choice of a zero point. Therefore, only relative free energy values, or changes in free energy, are physically meaningful.
In electrochemistry, the Nernst equation is a chemical thermodynamical relationship that permits the calculation of the reduction potential of a reaction from the standard electrode potential, absolute temperature, the number of electrons involved in the redox reaction, and activities of the chemical species undergoing reduction and oxidation respectively. It was named after Walther Nernst, a German physical chemist who formulated the equation.
In chemistry, the standard molar entropy is the entropy content of one mole of pure substance at a standard state of pressure and any temperature of interest. These are often chosen to be the standard temperature and pressure.
In thermodynamics, the chemical potential of a species is the energy that can be absorbed or released due to a change of the particle number of the given species, e.g. in a chemical reaction or phase transition. The chemical potential of a species in a mixture is defined as the rate of change of free energy of a thermodynamic system with respect to the change in the number of atoms or molecules of the species that are added to the system. Thus, it is the partial derivative of the free energy with respect to the amount of the species, all other species' concentrations in the mixture remaining constant. When both temperature and pressure are held constant, and the number of particles is expressed in moles, the chemical potential is the partial molar Gibbs free energy. At chemical equilibrium or in phase equilibrium, the total sum of the product of chemical potentials and stoichiometric coefficients is zero, as the free energy is at a minimum. In a system in diffusion equilibrium, the chemical potential of any chemical species is uniformly the same everywhere throughout the system.
A thermodynamic potential is a scalar quantity used to represent the thermodynamic state of a system. Just as in mechanics, where potential energy is defined as capacity to do work, similarly different potentials have different meanings. The concept of thermodynamic potentials was introduced by Pierre Duhem in 1886. Josiah Willard Gibbs in his papers used the term fundamental functions. While thermodynamic potentials cannot be measured directly, they can be predicted using computational chemistry.
In thermodynamics, the Helmholtz free energy is a thermodynamic potential that measures the useful work obtainable from a closed thermodynamic system at a constant temperature (isothermal). The change in the Helmholtz energy during a process is equal to the maximum amount of work that the system can perform in a thermodynamic process in which temperature is held constant. At constant temperature, the Helmholtz free energy is minimized at equilibrium.
In physics, a partition function describes the statistical properties of a system in thermodynamic equilibrium. Partition functions are functions of the thermodynamic state variables, such as the temperature and volume. Most of the aggregate thermodynamic variables of the system, such as the total energy, free energy, entropy, and pressure, can be expressed in terms of the partition function or its derivatives. The partition function is dimensionless.
The standard enthalpy of reaction for a chemical reaction is the difference between total product and total reactant molar enthalpies, calculated for substances in their standard states. The value can be approximately interpreted in terms of the total of the chemical bond energies for bonds broken and bonds formed.
In chemical thermodynamics, an endergonic reaction is a chemical reaction in which the standard change in free energy is positive, and an additional driving force is needed to perform this reaction. In layman's terms, the total amount of useful energy is negative so the total energy is a net negative result, as opposed to a net positive result in an exergonic reaction. Another way to phrase this is that useful energy must be absorbed from the surroundings into the workable system for the reaction to happen.
The equilibrium constant of a chemical reaction is the value of its reaction quotient at chemical equilibrium, a state approached by a dynamic chemical system after sufficient time has elapsed at which its composition has no measurable tendency towards further change. For a given set of reaction conditions, the equilibrium constant is independent of the initial analytical concentrations of the reactant and product species in the mixture. Thus, given the initial composition of a system, known equilibrium constant values can be used to determine the composition of the system at equilibrium. However, reaction parameters like temperature, solvent, and ionic strength may all influence the value of the equilibrium constant.
In chemical thermodynamics, the reaction quotient (Qr or just Q) is a dimensionless quantity that provides a measurement of the relative amounts of products and reactants present in a reaction mixture for a reaction with well-defined overall stoichiometry at a particular point in time. Mathematically, it is defined as the ratio of the activities (or molar concentrations) of the product species over those of the reactant species involved in the chemical reaction, taking stoichiometric coefficients of the reaction into account as exponents of the concentrations. In equilibrium, the reaction quotient is constant over time and is equal to the equilibrium constant.
The Van 't Hoff equation relates the change in the equilibrium constant, Keq, of a chemical reaction to the change in temperature, T, given the standard enthalpy change, ΔrH⊖, for the process. The subscript means "reaction" and the superscript means "standard". It was proposed by Dutch chemist Jacobus Henricus van 't Hoff in 1884 in his book Études de Dynamique chimique.
In thermodynamics, the fundamental thermodynamic relation are four fundamental equations which demonstrate how four important thermodynamic quantities depend on variables that can be controlled and measured experimentally. Thus, they are essentially equations of state, and using the fundamental equations, experimental data can be used to determine sought-after quantities like G or H (enthalpy). The relation is generally expressed as a microscopic change in internal energy in terms of microscopic changes in entropy, and volume for a closed system in thermal equilibrium in the following way.
Thermodynamic databases contain information about thermodynamic properties for substances, the most important being enthalpy, entropy, and Gibbs free energy. Numerical values of these thermodynamic properties are collected as tables or are calculated from thermodynamic datafiles. Data is expressed as temperature-dependent values for one mole of substance at the standard pressure of 101.325 kPa, or 100 kPa. Both of these definitions for the standard condition for pressure are in use.
In chemistry, transition state theory (TST) explains the reaction rates of elementary chemical reactions. The theory assumes a special type of chemical equilibrium (quasi-equilibrium) between reactants and activated transition state complexes.
Equilibrium chemistry is concerned with systems in chemical equilibrium. The unifying principle is that the free energy of a system at equilibrium is the minimum possible, so that the slope of the free energy with respect to the reaction coordinate is zero. This principle, applied to mixtures at equilibrium provides a definition of an equilibrium constant. Applications include acid–base, host–guest, metal–complex, solubility, partition, chromatography and redox equilibria.
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.