In chemistry, a nucleophile is a chemical species that forms bonds by donating an electron pair. All molecules and ions with a free pair of electrons or at least one pi bond can act as nucleophiles. Because nucleophiles donate electrons, they are Lewis bases.

For organic chemistry, a carbonyl group is a functional group with the formula C=O, composed of a carbon atom double-bonded to an oxygen atom, and it is divalent at the C atom. It is common to several classes of organic compounds, as part of many larger functional groups. A compound containing a carbonyl group is often referred to as a carbonyl compound.
In chemistry, a nucleophilic substitution (SN) is a class of chemical reactions in which an electron-rich chemical species replaces a functional group within another electron-deficient molecule. The molecule that contains the electrophile and the leaving functional group is called the substrate.
In chemistry, an electrophile is a chemical species that forms bonds with nucleophiles by accepting an electron pair. Because electrophiles accept electrons, they are Lewis acids. Most electrophiles are positively charged, have an atom that carries a partial positive charge, or have an atom that does not have an octet of electrons.
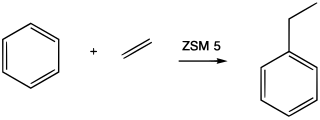
Alkylation is a chemical reaction that entails transfer of an alkyl group. The alkyl group may be transferred as an alkyl carbocation, a free radical, a carbanion, or a carbene. Alkylating agents are reagents for effecting alkylation. Alkyl groups can also be removed in a process known as dealkylation. Alkylating agents are often classified according to their nucleophilic or electrophilic character. In oil refining contexts, alkylation refers to a particular alkylation of isobutane with olefins. For upgrading of petroleum, alkylation produces a premium blending stock for gasoline. In medicine, alkylation of DNA is used in chemotherapy to damage the DNA of cancer cells. Alkylation is accomplished with the class of drugs called alkylating antineoplastic agents.

In organometallic chemistry, organolithium reagents are chemical compounds that contain carbon–lithium (C–Li) bonds. These reagents are important in organic synthesis, and are frequently used to transfer the organic group or the lithium atom to the substrates in synthetic steps, through nucleophilic addition or simple deprotonation. Organolithium reagents are used in industry as an initiator for anionic polymerization, which leads to the production of various elastomers. They have also been applied in asymmetric synthesis in the pharmaceutical industry. Due to the large difference in electronegativity between the carbon atom and the lithium atom, the C−Li bond is highly ionic. Owing to the polar nature of the C−Li bond, organolithium reagents are good nucleophiles and strong bases. For laboratory organic synthesis, many organolithium reagents are commercially available in solution form. These reagents are highly reactive, and are sometimes pyrophoric.
A substitution reaction is a chemical reaction during which one functional group in a chemical compound is replaced by another functional group. Substitution reactions are of prime importance in organic chemistry. Substitution reactions in organic chemistry are classified either as electrophilic or nucleophilic depending upon the reagent involved, whether a reactive intermediate involved in the reaction is a carbocation, a carbanion or a free radical, and whether the substrate is aliphatic or aromatic. Detailed understanding of a reaction type helps to predict the product outcome in a reaction. It also is helpful for optimizing a reaction with regard to variables such as temperature and choice of solvent.
Electrophilic substitution reactions are chemical reactions in which an electrophile displaces a functional group in a compound, which is typically, but not always, aromatic. Aromatic substitution reactions are characteristic of aromatic compounds and are common ways of introducing functional groups into benzene rings. Some aliphatic compounds can undergo electrophilic substitution as well.

In organic chemistry, the Michael reaction or Michael 1,4 addition is a reaction between a Michael donor and a Michael acceptor to produce a Michael adduct by creating a carbon-carbon bond at the acceptor's β-carbon. It belongs to the larger class of conjugate additions and is widely used for the mild formation of carbon-carbon bonds.
A tetrahedral intermediate is a reaction intermediate in which the bond arrangement around an initially double-bonded carbon atom has been transformed from trigonal to tetrahedral. Tetrahedral intermediates result from nucleophilic addition to a carbonyl group. The stability of tetrahedral intermediate depends on the ability of the groups attached to the new tetrahedral carbon atom to leave with the negative charge. Tetrahedral intermediates are very significant in organic syntheses and biological systems as a key intermediate in esterification, transesterification, ester hydrolysis, formation and hydrolysis of amides and peptides, hydride reductions, and other chemical reactions.
In organic chemistry, Madelung synthesis is a chemical reaction that produces indoles by the intramolecular cyclization of N-phenylamides using strong base at high temperature. The Madelung synthesis was reported in 1912 by Walter Madelung, when he observed that 2-phenylindole was synthesized using N-benzoyl-o-toluidine and two equivalents of sodium ethoxide in a heated, airless reaction. Common reaction conditions include use of sodium or potassium alkoxide as base in hexane or tetrahydrofuran solvents, at temperatures ranging between 200–400 °C. A hydrolysis step is also required in the synthesis. The Madelung synthesis is important because it is one of few known reactions that produce indoles from a base-catalyzed thermal cyclization of N-acyl-o-toluidines.

Nucleophilic conjugate addition is a type of organic reaction. Ordinary nucleophilic additions or 1,2-nucleophilic additions deal mostly with additions to carbonyl compounds. Simple alkene compounds do not show 1,2 reactivity due to lack of polarity, unless the alkene is activated with special substituents. With α,β-unsaturated carbonyl compounds such as cyclohexenone it can be deduced from resonance structures that the β position is an electrophilic site which can react with a nucleophile. The negative charge in these structures is stored as an alkoxide anion. Such a nucleophilic addition is called a nucleophilic conjugate addition or 1,4-nucleophilic addition. The most important active alkenes are the aforementioned conjugated carbonyls and acrylonitriles.
In organic chemistry, umpolung or polarity inversion is the chemical modification of a functional group with the aim of the reversal of polarity of that group. This modification allows secondary reactions of this functional group that would otherwise not be possible. The concept was introduced by D. Seebach and E.J. Corey. Polarity analysis during retrosynthetic analysis tells a chemist when umpolung tactics are required to synthesize a target molecule.

Asymmetric induction describes the preferential formation in a chemical reaction of one enantiomer or diastereoisomer over the other as a result of the influence of a chiral feature present in the substrate, reagent, catalyst or environment. Asymmetric induction is a key element in asymmetric synthesis.

The Bürgi–Dunitz angle is one of two angles that fully define the geometry of "attack" of a nucleophile on a trigonal unsaturated center in a molecule, originally the carbonyl center in an organic ketone, but now extending to aldehyde, ester, and amide carbonyls, and to alkenes (olefins) as well. The angle was named after crystallographers Hans-Beat Bürgi and Jack D. Dunitz, its first senior investigators.
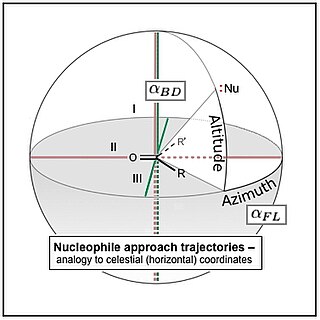
The Flippin–Lodge angle is one of two angles used by organic and biological chemists studying the relationship between a molecule's chemical structure and ways that it reacts, for reactions involving "attack" of an electron-rich reacting species, the nucleophile, on an electron-poor reacting species, the electrophile. Specifically, the angles—the Bürgi–Dunitz, , and the Flippin–Lodge, —describe the "trajectory" or "angle of attack" of the nucleophile as it approaches the electrophile, in particular when the latter is planar in shape. This is called a nucleophilic addition reaction and it plays a central role in the biological chemistry taking place in many biosyntheses in nature, and is a central "tool" in the reaction toolkit of modern organic chemistry, e.g., to construct new molecules such as pharmaceuticals. Theory and use of these angles falls into the areas of synthetic and physical organic chemistry, which deals with chemical structure and reaction mechanism, and within a sub-specialty called structure correlation.

An oxocarbeniumion is a chemical species characterized by a central sp2-hybridized carbon, an oxygen substituent, and an overall positive charge that is delocalized between the central carbon and oxygen atoms. An oxocarbenium ion is represented by two limiting resonance structures, one in the form of a carbenium ion with the positive charge on carbon and the other in the form of an oxonium species with the formal charge on oxygen. As a resonance hybrid, the true structure falls between the two. Compared to neutral carbonyl compounds like ketones or esters, the carbenium ion form is a larger contributor to the structure. They are common reactive intermediates in the hydrolysis of glycosidic bonds, and are a commonly used strategy for chemical glycosylation. These ions have since been proposed as reactive intermediates in a wide range of chemical transformations, and have been utilized in the total synthesis of several natural products. In addition, they commonly appear in mechanisms of enzyme-catalyzed biosynthesis and hydrolysis of carbohydrates in nature. Anthocyanins are natural flavylium dyes, which are stabilized oxocarbenium compounds. Anthocyanins are responsible for the colors of a wide variety of common flowers such as pansies and edible plants such as eggplant and blueberry.
Vicinal difunctionalization refers to a chemical reaction involving transformations at two adjacent centers. This transformation can be accomplished in α,β-unsaturated carbonyl compounds via the conjugate addition of a nucleophile to the β-position followed by trapping of the resulting enolate with an electrophile at the α-position. When the nucleophile is an enolate and the electrophile a proton, the reaction is called Michael addition.
Electrophilic amination is a chemical process involving the formation of a carbon–nitrogen bond through the reaction of a nucleophilic carbanion with an electrophilic source of nitrogen.
Electrophilic substitution of unsaturated silanes involves attack of an electrophile on an allyl- or vinylsilane. An allyl or vinyl group is incorporated at the electrophilic center after loss of the silyl group.

