From alkenes using carbenoid reagents
Several methods exist for converting alkenes to cyclopropane rings using carbene type reagents. As carbenes themselves are highly reactive it is common for them to be used in a stabilised form, referred to as a carbenoid. [2]
Using diazo compounds
Certain diazo compounds, such as diazomethane, can react with olefins to produce cyclopropanes in a 2 step manner. The first step involves a 1,3-dipolar cycloaddition to form a pyrazoline which then undergoes denitrogenation, either photochemically or by thermal decomposition, to give cyclopropane. The thermal route, which often uses KOH and platinum as catalysts, is also known as the Kishner cyclopropane synthesis after the Russian chemist Nikolai Kischner [7] [8] and can also be performed using hydrazine and α,β-unsaturated carbonyl compounds. [9] The mechanism of decomposition has been the subject of several studies and remains somewhat controversial, although it is broadly thought to proceed via a diradical species. [10] [11] In terms of green chemistry this method is superior to other carbene based cyclopropanations; as it does not involve metals or halogenated reagents, and produces only N2 as a by-product. However the reaction can be dangerous as trace amounts of unreacted diazo compounds may explode during the thermal rearrangement of the pyrazoline.
-

Using free carbenes
Free carbenes can be employed for cyclopropanation reactions, however there is limited scope for this as few can be produced conveniently and nearly all are unstable (see: carbene dimerization). An exception are dihalocarbenes such as dichlorocarbene or difluorocarbene, which are reasonably stable and will react to form geminal dihalo-cyclopropanes. [15] These compounds can then be used to form allenes via the Skattebøl rearrangement.
-

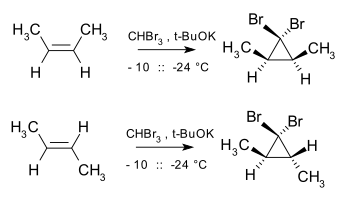
The Buchner ring expansion reaction also involves the formation of a stabilised carbene. Cyclopropanation is also stereospecific as the addition of carbene and carbenoids to alkenes is a form of a cheletropic reaction, with the addition taking place in a syn manner. For example, dibromocarbene and cis-2-butene yield cis-2,3-dimethyl-1,1-dibromocyclopropane, whereas the trans isomer exclusively yields the trans cyclopropane. [16]
-