An ylide or ylid is a neutral dipolar molecule containing a formally negatively charged atom (usually a carbanion) directly attached to a heteroatom with a formal positive charge (usually nitrogen, phosphorus or sulfur), and in which both atoms have full octets of electrons. The result can be viewed as a structure in which two adjacent atoms are connected by both a covalent and an ionic bond; normally written X+–Y−. Ylides are thus 1,2-dipolar compounds, and a subclass of zwitterions. They appear in organic chemistry as reagents or reactive intermediates.
Cyclopropene is an organic compound with the formula C3H4. It is the simplest cycloalkene. Because the ring is highly strained, cyclopropene is difficult to prepare and highly reactive. This colorless gas has been the subject for many fundamental studies of bonding and reactivity. It does not occur naturally, but derivatives are known in some fatty acids. Derivatives of cyclopropene are used commercially to control ripening of some fruit.
In organic chemistry, a carbene is a molecule containing a neutral carbon atom with a valence of two and two unshared valence electrons. The general formula is R−:C−R' or R=C: where the R represents substituents or hydrogen atoms.
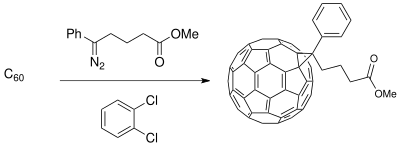
In organic chemistry, the diazo group is an organic moiety consisting of two linked nitrogen atoms at the terminal position. Overall charge-neutral organic compounds containing the diazo group bound to a carbon atom are called diazo compounds or diazoalkanes and are described by the general structural formula R2C=N+=N−. The simplest example of a diazo compound is diazomethane, CH2N2. Diazo compounds should not be confused with azo compounds or with diazonium compounds.
The 1,3-dipolar cycloaddition is a chemical reaction between a 1,3-dipole and a dipolarophile to form a five-membered ring. The earliest 1,3-dipolar cycloadditions were described in the late 19th century to the early 20th century, following the discovery of 1,3-dipoles. Mechanistic investigation and synthetic application were established in the 1960s, primarily through the work of Rolf Huisgen. Hence, the reaction is sometimes referred to as the Huisgen cycloaddition. 1,3-dipolar cycloaddition is an important route to the regio- and stereoselective synthesis of five-membered heterocycles and their ring-opened acyclic derivatives. The dipolarophile is typically an alkene or alkyne, but can be other pi systems. When the dipolarophile is an alkyne, aromatic rings are generally produced.
The Simmons–Smith reaction is an organic cheletropic reaction involving an organozinc carbenoid that reacts with an alkene to form a cyclopropane. It is named after Howard Ensign Simmons, Jr. and Ronald D. Smith. It uses a methylene free radical intermediate that is delivered to both carbons of the alkene simultaneously, therefore the configuration of the double bond is preserved in the product and the reaction is stereospecific.
An alkyne trimerisation is a [2+2+2] cycloaddition reaction in which three alkyne units react to form a benzene ring. The reaction requires a metal catalyst. The process is of historic interest as well as being applicable to organic synthesis. Being a cycloaddition reaction, it has high atom economy. Many variations have been developed, including cyclisation of mixtures of alkynes and alkenes as well as alkynes and nitriles.

The Bamford–Stevens reaction is a chemical reaction whereby treatment of tosylhydrazones with strong base gives alkenes. It is named for the British chemist William Randall Bamford and the Scottish chemist Thomas Stevens Stevens (1900–2000). The usage of aprotic solvents gives predominantly Z-alkenes, while protic solvent gives a mixture of E- and Z-alkenes. As an alkene-generating transformation, the Bamford–Stevens reaction has broad utility in synthetic methodology and complex molecule synthesis.
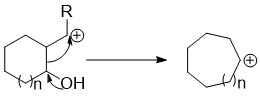
Ring expansion and ring contraction reactions expand or contract rings, usually in organic chemistry. The term usually refers to reactions involve making and breaking C-C bonds, Diverse mechanisms lead to these kinds of reactions.
Cycloheptatriene (CHT) is an organic compound with the formula C7H8. It is a closed ring of seven carbon atoms joined by three double bonds (as the name implies) and four single bonds. This colourless liquid has been of recurring theoretical interest in organic chemistry. It is a ligand in organometallic chemistry and a building block in organic synthesis. Cycloheptatriene is not aromatic, as reflected by the nonplanarity of the methylene bridge (-CH2-) with respect to the other atoms; however the related tropylium cation is.

The Johnson–Corey–Chaykovsky reaction is a chemical reaction used in organic chemistry for the synthesis of epoxides, aziridines, and cyclopropanes. It was discovered in 1961 by A. William Johnson and developed significantly by E. J. Corey and Michael Chaykovsky. The reaction involves addition of a sulfur ylide to a ketone, aldehyde, imine, or enone to produce the corresponding 3-membered ring. The reaction is diastereoselective favoring trans substitution in the product regardless of the initial stereochemistry. The synthesis of epoxides via this method serves as an important retrosynthetic alternative to the traditional epoxidation reactions of olefins.

The Wolff rearrangement is a reaction in organic chemistry in which an α-diazocarbonyl compound is converted into a ketene by loss of dinitrogen with accompanying 1,2-rearrangement. The Wolff rearrangement yields a ketene as an intermediate product, which can undergo nucleophilic attack with weakly acidic nucleophiles such as water, alcohols, and amines, to generate carboxylic acid derivatives or undergo [2+2] cycloaddition reactions to form four-membered rings. The mechanism of the Wolff rearrangement has been the subject of debate since its first use. No single mechanism sufficiently describes the reaction, and there are often competing concerted and carbene-mediated pathways; for simplicity, only the textbook, concerted mechanism is shown below. The reaction was discovered by Ludwig Wolff in 1902. The Wolff rearrangement has great synthetic utility due to the accessibility of α-diazocarbonyl compounds, variety of reactions from the ketene intermediate, and stereochemical retention of the migrating group. However, the Wolff rearrangement has limitations due to the highly reactive nature of α-diazocarbonyl compounds, which can undergo a variety of competing reactions.

In organic chemistry, cyclopropanation refers to any chemical process which generates cyclopropane rings. It is an important process in modern chemistry as many useful compounds bear this motif; for example pyrethroid insecticides and a number of quinolone antibiotics. However, the high ring strain present in cyclopropanes makes them challenging to produce and generally requires the use of highly reactive species, such as carbenes, ylids and carbanions. Many of the reactions proceed in a cheletropic manner.

Carbene C−H insertion in organic chemistry concerns the insertion reaction of a carbene into a carbon–hydrogen bond. This organic reaction is of some importance in the synthesis of new organic compounds.
Intramolecular reactions of diazocarbonyl compounds include addition to carbon–carbon double bonds to form fused cyclopropanes and insertion into carbon–hydrogen bonds or carbon–carbon bonds.
Metal-catalyzed cyclopropanations are chemical reactions that result in the formation of a cyclopropane ring from a metal carbenoid species and an alkene. In the Simmons–Smith reaction the metal involved is zinc. Metal carbenoid species can be generated through the reaction of a diazo compound with a transition metal). The intramolecular variant of this reaction was first reported in 1961. Rhodium carboxylate complexes, such as dirhodium tetraacetate, are common catalysts. Enantioselective cyclopropanations have been developed.
Carbene dimerization is a type of organic reaction in which two carbene or carbenoid precursors react in a formal dimerization to an alkene. This reaction is often considered an unwanted side-reaction but it is also investigated as a synthetic tool. In this reaction type either the two carbenic intermediates react or a carbenic intermediate reacts with a carbene precursor. An early pioneer was Christoph Grundmann reporting on a carbene dimerisation in 1938. In the domain of persistent carbenes the Wanzlick equilibrium describes an equilibrium between a carbene and its alkene.

The Doyle–Kirmse reaction is an organic reaction in which a metal carbene reacts with an allyl compound with transposition of the alkene and transfer of the electronegative group from the allyl onto the carbene carbon.
Cycloisomerization is any isomerization in which the cyclic isomer of the substrate is produced in the reaction coordinate. The greatest advantage of cycloisomerization reactions is its atom economical nature, by design nothing is wasted, as every atom in the starting material is present in the product. In most cases these reactions are mediated by a transition metal catalyst, in few cases organocatalysts and rarely do they occur under thermal conditions. These cyclizations are able to be performed with excellent levels of selectivity in numerous cases and have transformed cycloisomerization into a powerful tool for unique and complex molecular construction. Cycloisomerization is a very broad topic in organic synthesis and many reactions that would be categorized as such exist. Two basic classes of these reactions are intramolecular Michael addition and Intramolecular Diels–Alder reactions. Under the umbrella of cycloisomerization, enyne and related olefin cycloisomerizations are the most widely used and studied reactions.
Cobalt(II)–porphyrin catalysis is a process in which a Co(II) porphyrin complex acts as a catalyst, inducing and accelerating a chemical reaction.