The Friedel–Crafts reactions are a set of reactions developed by Charles Friedel and James Crafts in 1877 to attach substituents to an aromatic ring. Friedel–Crafts reactions are of two main types: alkylation reactions and acylation reactions. Both proceed by electrophilic aromatic substitution.

In organic chemistry, the ene reaction is a chemical reaction between an alkene with an allylic hydrogen and a compound containing a multiple bond, in order to form a new σ-bond with migration of the ene double bond and 1,5 hydrogen shift. The product is a substituted alkene with the double bond shifted to the allylic position.
A 1,2-rearrangement or 1,2-migration or 1,2-shift or Whitmore 1,2-shift is an organic reaction where a substituent moves from one atom to another atom in a chemical compound. In a 1,2 shift the movement involves two adjacent atoms but moves over larger distances are possible. In the example below the substituent R moves from carbon atom C2 to C3.
A cascade reaction, also known as a domino reaction or tandem reaction, is a chemical process that comprises at least two consecutive reactions such that each subsequent reaction occurs only in virtue of the chemical functionality formed in the previous step. In cascade reactions, isolation of intermediates is not required, as each reaction composing the sequence occurs spontaneously. In the strictest definition of the term, the reaction conditions do not change among the consecutive steps of a cascade and no new reagents are added after the initial step. By contrast, one-pot procedures similarly allow at least two reactions to be carried out consecutively without any isolation of intermediates, but do not preclude the addition of new reagents or the change of conditions after the first reaction. Thus, any cascade reaction is also a one-pot procedure, while the reverse does not hold true. Although often composed solely of intramolecular transformations, cascade reactions can also occur intermolecularly, in which case they also fall under the category of multicomponent reactions.
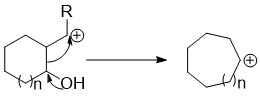
Ring expansion and ring contraction reactions expand or contract rings, usually in organic chemistry. The term usually refers to reactions involve making and breaking C-C bonds, Diverse mechanisms lead to these kinds of reactions.

The Baker–Venkataraman rearrangement is the chemical reaction of 2-acetoxyacetophenones with base to form 1,3-diketones.

The Dakin oxidation (or Dakin reaction) is an organic redox reaction in which an ortho- or para-hydroxylated phenyl aldehyde (2-hydroxybenzaldehyde or 4-hydroxybenzaldehyde) or ketone reacts with hydrogen peroxide (H2O2) in base to form a benzenediol and a carboxylate. Overall, the carbonyl group is oxidised, whereas the H2O2 is reduced.

The Tiffeneau–Demjanov rearrangement is the chemical reaction of a 1-aminomethyl-cycloalkanol with nitrous acid to form an enlarged cycloketone.
The Gabriel–Colman rearrangement is the chemical reaction of a saccharin or phthalimido ester with a strong base, such as an alkoxide, to form substituted isoquinolines. First described in 1900 by chemists Siegmund Gabriel and James Colman, this rearrangement, a ring expansion, is seen to be general if there is an enolizable hydrogen on the group attached to the nitrogen, since it is necessary for the nitrogen to abstract a hydrogen to form the carbanion that will close the ring. As shown in the case of the general example below, X is either CO or SO2.

The Wolff rearrangement is a reaction in organic chemistry in which an α-diazocarbonyl compound is converted into a ketene by loss of dinitrogen with accompanying 1,2-rearrangement. The Wolff rearrangement yields a ketene as an intermediate product, which can undergo nucleophilic attack with weakly acidic nucleophiles such as water, alcohols, and amines, to generate carboxylic acid derivatives or undergo [2+2] cycloaddition reactions to form four-membered rings. The mechanism of the Wolff rearrangement has been the subject of debate since its first use. No single mechanism sufficiently describes the reaction, and there are often competing concerted and carbene-mediated pathways; for simplicity, only the textbook, concerted mechanism is shown below. The reaction was discovered by Ludwig Wolff in 1902. The Wolff rearrangement has great synthetic utility due to the accessibility of α-diazocarbonyl compounds, variety of reactions from the ketene intermediate, and stereochemical retention of the migrating group. However, the Wolff rearrangement has limitations due to the highly reactive nature of α-diazocarbonyl compounds, which can undergo a variety of competing reactions.
The Nazarov cyclization reaction is a chemical reaction used in organic chemistry for the synthesis of cyclopentenones. The reaction is typically divided into classical and modern variants, depending on the reagents and substrates employed. It was originally discovered by Ivan Nikolaevich Nazarov (1906–1957) in 1941 while studying the rearrangements of allyl vinyl ketones.
The Rubottom oxidation is a useful, high-yielding chemical reaction between silyl enol ethers and peroxyacids to give the corresponding α-hydroxy carbonyl product. The mechanism of the reaction was proposed in its original disclosure by A.G. Brook with further evidence later supplied by George M. Rubottom. After a Prilezhaev-type oxidation of the silyl enol ether with the peroxyacid to form the siloxy oxirane intermediate, acid-catalyzed ring-opening yields an oxocarbenium ion. This intermediate then participates in a 1,4-silyl migration to give an α-siloxy carbonyl derivative that can be readily converted to the α-hydroxy carbonyl compound in the presence of acid, base, or a fluoride source.
The Meyer–Schuster rearrangement is the chemical reaction described as an acid-catalyzed rearrangement of secondary and tertiary propargyl alcohols to α,β-unsaturated ketones if the alkyne group is internal and α,β-unsaturated aldehydes if the alkyne group is terminal. Reviews have been published by Swaminathan and Narayan, Vartanyan and Banbanyan, and Engel and Dudley, the last of which describes ways to promote the Meyer–Schuster rearrangement over other reactions available to propargyl alcohols.
The Stieglitz rearrangement is a rearrangement reaction in organic chemistry which is named after the American chemist Julius Stieglitz (1867–1937) and was first investigated by him and Paul Nicholas Leech in 1913. It describes the 1,2-rearrangement of trityl amine derivatives to triaryl imines. It is comparable to a Beckmann rearrangement which also involves a substitution at a nitrogen atom through a carbon to nitrogen shift. As an example, triaryl hydroxylamines can undergo a Stieglitz rearrangement by dehydration and the shift of a phenyl group after activation with phosphorus pentachloride to yield the respective triaryl imine, a Schiff base.
Selenoxide elimination is a method for the chemical synthesis of alkenes from selenoxides. It is most commonly used to synthesize α,β-unsaturated carbonyl compounds from the corresponding saturated analogues. It is mechanistically related to the Cope reaction.
Melengestrol is a steroidal progestin of the 17α-hydroxyprogesterone group and an antineoplastic drug which was never marketed. An acylated derivative, melengestrol acetate, is used as a growth promoter in animals.

An oxaziridine is an organic molecule that features a three-membered heterocycle containing oxygen, nitrogen, and carbon. In their largest application, oxaziridines are intermediates in the industrial production of hydrazine. Oxaziridine derivatives are also used as specialized reagents in organic chemistry for a variety of oxidations, including alpha hydroxylation of enolates, epoxidation and aziridination of olefins, and other heteroatom transfer reactions. Oxaziridines also serve as precursors to nitrones and participate in [3+2] cycloadditions with various heterocumulenes to form substituted five-membered heterocycles. Chiral oxaziridine derivatives effect asymmetric oxygen transfer to prochiral enolates as well as other substrates. Some oxaziridines also have the property of a high barrier to inversion of the nitrogen, allowing for the possibility of chirality at the nitrogen center.
The Buchner ring expansion is a two-step organic C-C bond forming reaction used to access 7-membered rings. The first step involves formation of a carbene from ethyl diazoacetate, which cyclopropanates an aromatic ring. The ring expansion occurs in the second step, with an electrocyclic reaction opening the cyclopropane ring to form the 7-membered ring.
In organic chemistry, Lewis acid catalysis is the use of metal-based Lewis acids as catalysts for organic reactions. The acids act as an electron pair acceptor to increase the reactivity of a substrate. Common Lewis acid catalysts are based on main group metals such as aluminum, boron, silicon, and tin, as well as many early and late d-block metals. The metal atom forms an adduct with a lone-pair bearing electronegative atom in the substrate, such as oxygen, nitrogen, sulfur, and halogens. The complexation has partial charge-transfer character and makes the lone-pair donor effectively more electronegative, activating the substrate toward nucleophilic attack, heterolytic bond cleavage, or cycloaddition with 1,3-dienes and 1,3-dipoles.
Cycloisomerization is any isomerization in which the cyclic isomer of the substrate is produced in the reaction coordinate. The greatest advantage of cycloisomerization reactions is its atom economical nature, by design nothing is wasted, as every atom in the starting material is present in the product. In most cases these reactions are mediated by a transition metal catalyst, in few cases organocatalysts and rarely do they occur under thermal conditions. These cyclizations are able to be performed with excellent levels of selectivity in numerous cases and have transformed cycloisomerization into a powerful tool for unique and complex molecular construction. Cycloisomerization is a very broad topic in organic synthesis and many reactions that would be categorized as such exist. Two basic classes of these reactions are intramolecular Michael addition and Intramolecular Diels–Alder reactions. Under the umbrella of cycloisomerization, enyne and related olefin cycloisomerizations are the most widely used and studied reactions.



