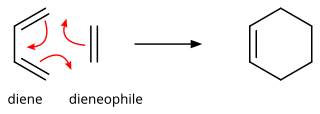
In organic chemistry, the Diels–Alder reaction is a chemical reaction between a conjugated diene and a substituted alkene, commonly termed the dienophile, to form a substituted cyclohexene derivative. It is the prototypical example of a pericyclic reaction with a concerted mechanism. More specifically, it is classified as a thermally allowed [4+2] cycloaddition with Woodward–Hoffmann symbol [π4s + π2s]. It was first described by Otto Diels and Kurt Alder in 1928. For the discovery of this reaction, they were awarded the Nobel Prize in Chemistry in 1950. Through the simultaneous construction of two new carbon–carbon bonds, the Diels–Alder reaction provides a reliable way to form six-membered rings with good control over the regio- and stereochemical outcomes. Consequently, it has served as a powerful and widely applied tool for the introduction of chemical complexity in the synthesis of natural products and new materials. The underlying concept has also been applied to π-systems involving heteroatoms, such as carbonyls and imines, which furnish the corresponding heterocycles; this variant is known as the hetero-Diels–Alder reaction. The reaction has also been generalized to other ring sizes, although none of these generalizations have matched the formation of six-membered rings in terms of scope or versatility. Because of the negative values of ΔH° and ΔS° for a typical Diels–Alder reaction, the microscopic reverse of a Diels–Alder reaction becomes favorable at high temperatures, although this is of synthetic importance for only a limited range of Diels–Alder adducts, generally with some special structural features; this reverse reaction is known as the retro-Diels–Alder reaction.

Photochemistry is the branch of chemistry concerned with the chemical effects of light. Generally, this term is used to describe a chemical reaction caused by absorption of ultraviolet, visible (400–750 nm), or infrared radiation (750–2500 nm).
In organic chemistry, a cycloaddition is a chemical reaction in which "two or more unsaturated molecules combine with the formation of a cyclic adduct in which there is a net reduction of the bond multiplicity". The resulting reaction is a cyclization reaction. Many but not all cycloadditions are concerted and thus pericyclic. Nonconcerted cycloadditions are not pericyclic. As a class of addition reaction, cycloadditions permit carbon–carbon bond formation without the use of a nucleophile or electrophile.
A sigmatropic reaction in organic chemistry is a pericyclic reaction wherein the net result is one σ-bond is changed to another σ-bond in an uncatalyzed intramolecular reaction. The name sigmatropic is the result of a compounding of the long-established sigma designation from single carbon–carbon bonds and the Greek word tropos, meaning turn. In this type of rearrangement reaction, a substituent moves from one part of a π-bonded system to another part in an intramolecular reaction with simultaneous rearrangement of the π system. True sigmatropic reactions are usually uncatalyzed, although Lewis acid catalysis is possible. Sigmatropic reactions often have transition-metal catalysts that form intermediates in analogous reactions. The most well-known of the sigmatropic rearrangements are the [3,3] Cope rearrangement, Claisen rearrangement, Carroll rearrangement, and the Fischer indole synthesis.
An electrocyclic reaction can either be classified as conrotatory or disrotatory based on the rotation at each end of the molecule. In conrotatory mode, both atomic orbitals of the end groups turn in the same direction. In disrotatory mode, the atomic orbitals of the end groups turn in opposite directions. The cis/trans geometry of the final product is directly decided by the difference between conrotation and disrotation.

In organic chemistry, cheletropic reactions, also known as chelotropic reactions, are a type of pericyclic reaction. Specifically, cheletropic reactions are a subclass of cycloadditions. The key distinguishing feature of cheletropic reactions is that on one of the reagents, both new bonds are being made to the same atom.

Azomethine ylides are nitrogen-based 1,3-dipoles, consisting of an iminium ion next to a carbanion. They are used in 1,3-dipolar cycloaddition reactions to form five-membered heterocycles, including pyrrolidines and pyrrolines. These reactions are highly stereo- and regioselective, and have the potential to form four new contiguous stereocenters. Azomethine ylides thus have high utility in total synthesis, and formation of chiral ligands and pharmaceuticals. Azomethine ylides can be generated from many sources, including aziridines, imines, and iminiums. They are often generated in situ, and immediately reacted with dipolarophiles.
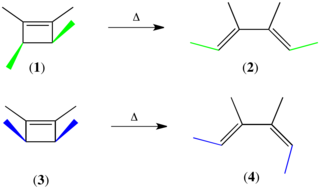
The Woodward–Hoffmann rules are a set of rules devised by Robert Burns Woodward and Roald Hoffmann to rationalize or predict certain aspects of the stereochemistry and activation energy of pericyclic reactions, an important class of reactions in organic chemistry. The rules originate in certain symmetries of the molecule's orbital structure that any molecular Hamiltonian conserves. Consequently, any symmetry-violating reaction must couple extensively to the environment; this imposes an energy barrier on its occurrence, and such reactions are called symmetry-forbidden. Their opposites are symmetry-allowed.
The Nazarov cyclization reaction is a chemical reaction used in organic chemistry for the synthesis of cyclopentenones. The reaction is typically divided into classical and modern variants, depending on the reagents and substrates employed. It was originally discovered by Ivan Nikolaevich Nazarov (1906–1957) in 1941 while studying the rearrangements of allyl vinyl ketones.
Organic photochemistry encompasses organic reactions that are induced by the action of light. The absorption of ultraviolet light by organic molecules often leads to reactions. In the earliest days, sunlight was employed, while in more modern times ultraviolet lamps are employed. Organic photochemistry has proven to be a very useful synthetic tool. Complex organic products can be obtained simply.
The vinylcyclopropane rearrangement or vinylcyclopropane-cyclopentene rearrangement is a ring expansion reaction, converting a vinyl-substituted cyclopropane ring into a cyclopentene ring.
In chemistry, frontier molecular orbital theory is an application of molecular orbital theory describing HOMO–LUMO interactions.
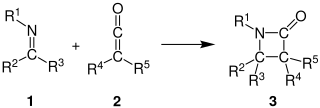
The Staudinger synthesis, also called the Staudinger ketene-imine cycloaddition, is a chemical synthesis in which an imine 1 reacts with a ketene 2 through a non-photochemical 2+2 cycloaddition to produce a β-lactam3. The reaction carries particular importance in the synthesis of β-lactam antibiotics. The Staudinger synthesis should not be confused with the Staudinger reaction, a phosphine or phosphite reaction used to reduce azides to amines.

Endiandric acid C, isolated from the tree Endiandra introrsa, is a well characterized chemical compound. Endiadric acid C is reported to have better antibiotic activity than ampicillin.

Torquoselectivity is a special kind of stereoselectivity observed in electrocyclic reactions in organic chemistry, defined as "the preference for inward or outward rotation of substituents in conrotatory or disrotatory electrocyclic reactions." Torquoselectivity is not to be confused with the normal diastereoselectivity seen in pericyclic reactions, as it represents a further level of selectivity beyond the Woodward-Hoffman rules. The name derives from the idea that the substituents in an electrocyclization appear to rotate over the course of the reaction, and thus selection of a single product is equivalent to selection of one direction of rotation. The concept was originally developed by Kendall N. Houk.
In 1976, the Italian chemist, Giovanni Piancatelli and coworkers developed a new method to synthesize 4-hydroxycyclopentenone derivatives from 2-furylcarbinols through an acid-catalyzed rearrangement. This discovery occurred when Piancatelli was studying heterocyclic steroids and their reactive abilities in an acidic environment. As this rearrangement has continued to be studied, it has become a commonly used rearrangement in natural product synthesis because of the ability to create 4-hydroxy-5-substitutedcyclopent-2-enones. Piancatelli’s motive for looking into this new rearrangement stemmed from the ever present 3-oxycyclopentene molecule, specifically its 5-hydroxy derivative, found in biologically active natural products.
The inverse electron demand Diels–Alder reaction, or DAINV or IEDDA is an organic chemical reaction, in which two new chemical bonds and a six-membered ring are formed. It is related to the Diels–Alder reaction, but unlike the Diels–Alder reaction, the DAINV is a cycloaddition between an electron-rich dienophile and an electron-poor diene. During a DAINV reaction, three pi-bonds are broken, and two sigma bonds and one new pi-bond are formed. A prototypical DAINV reaction is shown on the right.
The Buchner ring expansion is a two-step organic C-C bond forming reaction used to access 7-membered rings. The first step involves formation of a carbene from ethyl diazoacetate, which cyclopropanates an aromatic ring. The ring expansion occurs in the second step, with an electrocyclic reaction opening the cyclopropane ring to form the 7-membered ring.
The [4+4] Photocycloaddition is a cycloaddition reaction in which two unsaturated molecules connect via four atoms from each molecule to create an eight-membered ring. As a photochemical reaction, it is promoted by some form of light, as opposed to a thermal process.
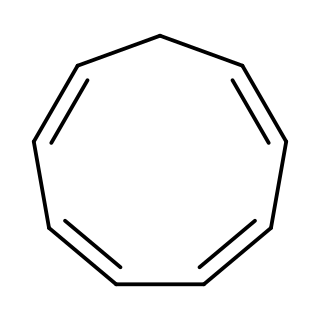
Cyclononatetraene is an organic compound with the formula C9H10. It was first prepared in 1969 by protonation of the corresponding aromatic anion (described below). It is unstable and isomerizes with a half-life of 50 minutes at room temperature to 3a,7a-dihydro-1H-indene via a thermal 6π disrotatory electrocyclic ring closing. Upon exposure to ultraviolet light, it undergoes a photochemical 8π electrocyclic ring closing to give bicyclo[6.1.0]nona-2,4,6-triene.











