
The aldol reaction is a reaction in organic chemistry that combines two carbonyl compounds to form a new β-hydroxy carbonyl compound. Its simplest form might involve the nucleophilic addition of an enolized ketone to another:

In organometallic chemistry, organolithium reagents are chemical compounds that contain carbon–lithium (C–Li) bonds. These reagents are important in organic synthesis, and are frequently used to transfer the organic group or the lithium atom to the substrates in synthetic steps, through nucleophilic addition or simple deprotonation. Organolithium reagents are used in industry as an initiator for anionic polymerization, which leads to the production of various elastomers. They have also been applied in asymmetric synthesis in the pharmaceutical industry. Due to the large difference in electronegativity between the carbon atom and the lithium atom, the C−Li bond is highly ionic. Owing to the polar nature of the C−Li bond, organolithium reagents are good nucleophiles and strong bases. For laboratory organic synthesis, many organolithium reagents are commercially available in solution form. These reagents are highly reactive, and are sometimes pyrophoric.
In organic chemistry, an electrocyclic reaction is a type of pericyclic rearrangement where the net result is one pi bond being converted into one sigma bond or vice versa. These reactions are usually categorized by the following criteria:

In organic chemistry, the Michael reaction or Michael 1,4 addition is a reaction between a Michael donor and a Michael acceptor to produce a Michael adduct by creating a carbon-carbon bond at the acceptor's β-carbon. It belongs to the larger class of conjugate additions and is widely used for the mild formation of carbon-carbon bonds.
The Robinson annulation is a chemical reaction used in organic chemistry for ring formation. It was discovered by Robert Robinson in 1935 as a method to create a six membered ring by forming three new carbon–carbon bonds. The method uses a ketone and a methyl vinyl ketone to form an α,β-unsaturated ketone in a cyclohexane ring by a Michael addition followed by an aldol condensation. This procedure is one of the key methods to form fused ring systems.
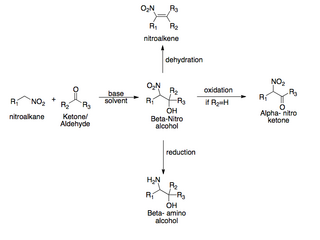
The Henry reaction is a classic carbon–carbon bond formation reaction in organic chemistry. Discovered in 1895 by the Belgian chemist Louis Henry (1834–1913), it is the combination of a nitroalkane and an aldehyde or ketone in the presence of a base to form β-nitro alcohols. This type of reaction is also referred to as a nitroaldol reaction. It is nearly analogous to the aldol reaction that had been discovered 23 years prior that couples two carbonyl compounds to form β-hydroxy carbonyl compounds known as "aldols". The Henry reaction is a useful technique in the area of organic chemistry due to the synthetic utility of its corresponding products, as they can be easily converted to other useful synthetic intermediates. These conversions include subsequent dehydration to yield nitroalkenes, oxidation of the secondary alcohol to yield α-nitro ketones, or reduction of the nitro group to yield β-amino alcohols.

In stereochemistry, a chiral auxiliary is a stereogenic group or unit that is temporarily incorporated into an organic compound in order to control the stereochemical outcome of the synthesis. The chirality present in the auxiliary can bias the stereoselectivity of one or more subsequent reactions. The auxiliary can then be typically recovered for future use.

The Fujimoto–Belleau reaction is a chemical reaction that forms cyclic α-substituted α,β-unsaturated ketones from enol lactones. The reaction was discovered in 1951 by George I. Fujimoto and Bernard Belleau. Belleau used this reaction to synthesize 1-methyl-3-keto-1,2,3,9,10,10a-hexahydrophenanthrene from a ketoacid starting material and Fujimoto demonstrated that steroids could be synthesized from naturally occurring lactone species using this method as well.
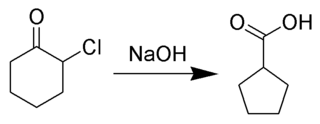
The Favorskii rearrangement is principally a rearrangement of cyclopropanones and α-halo ketones that leads to carboxylic acid derivatives. In the case of cyclic α-halo ketones, the Favorskii rearrangement constitutes a ring contraction. This rearrangement takes place in the presence of a base, sometimes hydroxide, to yield a carboxylic acid, but usually either an alkoxide base or an amine to yield an ester or an amide, respectively. α,α'-Dihaloketones eliminate HX under the reaction conditions to give α,β-unsaturated carbonyl compounds. Note that trihalomethyl ketone substrates will result in haloform and carboxylate formation via the haloform reaction instead.

The Wolff rearrangement is a reaction in organic chemistry in which an α-diazocarbonyl compound is converted into a ketene by loss of dinitrogen with accompanying 1,2-rearrangement. The Wolff rearrangement yields a ketene as an intermediate product, which can undergo nucleophilic attack with weakly acidic nucleophiles such as water, alcohols, and amines, to generate carboxylic acid derivatives or undergo [2+2] cycloaddition reactions to form four-membered rings. The mechanism of the Wolff rearrangement has been the subject of debate since its first use. No single mechanism sufficiently describes the reaction, and there are often competing concerted and carbene-mediated pathways; for simplicity, only the textbook, concerted mechanism is shown below. The reaction was discovered by Ludwig Wolff in 1902. The Wolff rearrangement has great synthetic utility due to the accessibility of α-diazocarbonyl compounds, variety of reactions from the ketene intermediate, and stereochemical retention of the migrating group. However, the Wolff rearrangement has limitations due to the highly reactive nature of α-diazocarbonyl compounds, which can undergo a variety of competing reactions.
The Rubottom oxidation is a useful, high-yielding chemical reaction between silyl enol ethers and peroxyacids to give the corresponding α-hydroxy carbonyl product. The mechanism of the reaction was proposed in its original disclosure by A.G. Brook with further evidence later supplied by George M. Rubottom. After a Prilezhaev-type oxidation of the silyl enol ether with the peroxyacid to form the siloxy oxirane intermediate, acid-catalyzed ring-opening yields an oxocarbenium ion. This intermediate then participates in a 1,4-silyl migration to give an α-siloxy carbonyl derivative that can be readily converted to the α-hydroxy carbonyl compound in the presence of acid, base, or a fluoride source.
In organic chemistry, aldol reactions are acid- or base-catalyzed reactions of aldehydes or ketones.
A (4+3) cycloaddition is a cycloaddition between a four-atom π-system and a three-atom π-system to form a seven-membered ring. Allyl or oxyallyl cations (propenylium-2-olate) are commonly used three-atom π-systems, while a diene plays the role of the four-atom π-system. It represents one of the relatively few synthetic methods available to form seven-membered rings stereoselectively in high yield.
In organic chemistry, α-halo ketones can be reduced with loss of the halogen atom to form enolates. The α-halo ketones are readily prepared from ketones by various ketone halogenation reactions, and the products are reactive intermediates that can be used for a variety of other chemical reactions.
2-Cyclopentenone is the organic compound with the chemical formula (CH2)2(CH)2CO. 2-Cyclopentenone contains two functional groups, a ketone and an alkene. It is a colorless liquid. Its isomer, 3-cyclopentenone is less commonly encountered.
In organic chemistry, the Baylis–Hillman, Morita–Baylis–Hillman, or MBH reaction is a carbon-carbon bond-forming reaction between an activated alkene and a carbon electrophile in the presence of a nucleophilic catalyst, such as a tertiary amine or phosphine. The product is densely functionalized, joining the alkene at the α-position to a reduced form of the electrophile.

Torquoselectivity is a special kind of stereoselectivity observed in electrocyclic reactions in organic chemistry, defined as "the preference for inward or outward rotation of substituents in conrotatory or disrotatory electrocyclic reactions." Torquoselectivity is not to be confused with the normal diastereoselectivity seen in pericyclic reactions, as it represents a further level of selectivity beyond the Woodward-Hoffman rules. The name derives from the idea that the substituents in an electrocyclization appear to rotate over the course of the reaction, and thus selection of a single product is equivalent to selection of one direction of rotation. The concept was originally developed by Kendall N. Houk.

Hydrogen-bond catalysis is a type of organocatalysis that relies on use of hydrogen bonding interactions to accelerate and control organic reactions. In biological systems, hydrogen bonding plays a key role in many enzymatic reactions, both in orienting the substrate molecules and lowering barriers to reaction. The field is relatively undeveloped compared to research in Lewis acid catalysis.
Rearrangements, especially those that can participate in cascade reactions, such as the aza-Cope rearrangements, are of high practical as well as conceptual importance in organic chemistry, due to their ability to quickly build structural complexity out of simple starting materials. The aza-Cope rearrangements are examples of heteroatom versions of the Cope rearrangement, which is a [3,3]-sigmatropic rearrangement that shifts single and double bonds between two allylic components. In accordance with the Woodward-Hoffman rules, thermal aza-Cope rearrangements proceed suprafacially. Aza-Cope rearrangements are generally classified by the position of the nitrogen in the molecule :
In organic chemistry, the Conia-ene reaction is an intramolecular cyclization reaction between an enolizable carbonyl such as an ester or ketone and an alkyne or alkene, giving a cyclic product with a new carbon-carbon bond. As initially reported by J. M. Conia and P. Le Perchec, the Conia-ene reaction is a heteroatom analog of the ene reaction that uses an enol as the ene component. Like other pericyclic reactions, the original Conia-ene reaction required high temperatures to proceed, limiting its wider application. However, subsequent improvements, particularly in metal catalysis, have led to significant expansion of reaction scope. Consequently, various forms of the Conia-ene reaction have been employed in the synthesis of complex molecules and natural products.






















