A chemical reaction is a process that leads to the chemical transformation of one set of chemical substances to another. When chemical reactions occur, the atoms are rearranged and the reaction is accompanied by an energy change as new products are generated. Classically, chemical reactions encompass changes that only involve the positions of electrons in the forming and breaking of chemical bonds between atoms, with no change to the nuclei, and can often be described by a chemical equation. Nuclear chemistry is a sub-discipline of chemistry that involves the chemical reactions of unstable and radioactive elements where both electronic and nuclear changes can occur.

Organic reactions are chemical reactions involving organic compounds. The basic organic chemistry reaction types are addition reactions, elimination reactions, substitution reactions, pericyclic reactions, rearrangement reactions, photochemical reactions and redox reactions. In organic synthesis, organic reactions are used in the construction of new organic molecules. The production of many man-made chemicals such as drugs, plastics, food additives, fabrics depend on organic reactions.

In organic chemistry, aromaticity is a chemical property describing the way in which a conjugated ring of unsaturated bonds, lone pairs, or empty orbitals exhibits a stabilization stronger than would be expected by the stabilization of conjugation alone. The earliest use of the term was in an article by August Wilhelm Hofmann in 1855. There is no general relationship between aromaticity as a chemical property and the olfactory properties of such compounds.

Photochemistry is the branch of chemistry concerned with the chemical effects of light. Generally, this term is used to describe a chemical reaction caused by absorption of ultraviolet, visible (400–750 nm), or infrared radiation (750–2500 nm).
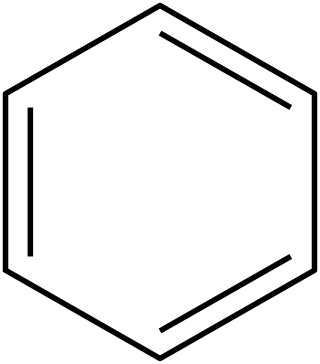
In organic chemistry, Hückel's rule predicts that a planar ring molecule will have aromatic properties if it has 4n + 2 π electrons, where n is a non-negative integer. The quantum mechanical basis for its formulation was first worked out by physical chemist Erich Hückel in 1931. The succinct expression as the 4n + 2 rule has been attributed to W. v. E. Doering (1951), although several authors were using this form at around the same time.
In organic chemistry, a cycloaddition is a chemical reaction in which "two or more unsaturated molecules combine with the formation of a cyclic adduct in which there is a net reduction of the bond multiplicity". The resulting reaction is a cyclization reaction. Many but not all cycloadditions are concerted and thus pericyclic. Nonconcerted cycloadditions are not pericyclic. As a class of addition reaction, cycloadditions permit carbon–carbon bond formation without the use of a nucleophile or electrophile.
A sigmatropic reaction in organic chemistry is a pericyclic reaction wherein the net result is one σ-bond is changed to another σ-bond in an uncatalyzed intramolecular reaction. The name sigmatropic is the result of a compounding of the long-established sigma designation from single carbon–carbon bonds and the Greek word tropos, meaning turn. In this type of rearrangement reaction, a substituent moves from one part of a π-bonded system to another part in an intramolecular reaction with simultaneous rearrangement of the π system. True sigmatropic reactions are usually uncatalyzed, although Lewis acid catalysis is possible. Sigmatropic reactions often have transition-metal catalysts that form intermediates in analogous reactions. The most well-known of the sigmatropic rearrangements are the [3,3] Cope rearrangement, Claisen rearrangement, Carroll rearrangement, and the Fischer indole synthesis.
In organic chemistry, a group transfer reaction is a class of the pericyclic reaction where one or more groups of atoms is transferred from one molecule to another. Group transfer reactions can sometimes be difficult to identify when separate reactant molecules combine into a single product molecule. Unlike other pericyclic reaction classes, group transfer reactions do not have a specific conversion of pi bonds into sigma bonds or vice versa, and tend to be less frequently encountered. Like all pericyclic reactions, group transfer reactions must obey the Woodward–Hoffmann rules. Group transfer reactions can be divided into two distinct subcategories: the ene reaction and the diimide reduction. Group transfer reactions have diverse applications in various fields, including protein adenylation, biocatalytic and chemoenzymatic approaches for chemical synthesis, and strengthening skim natural rubber latex.
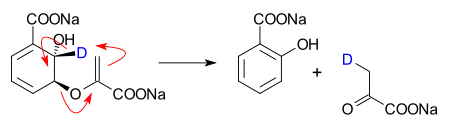
The 1,3-dipolar cycloaddition is a chemical reaction between a 1,3-dipole and a dipolarophile to form a five-membered ring. The earliest 1,3-dipolar cycloadditions were described in the late 19th century to the early 20th century, following the discovery of 1,3-dipoles. Mechanistic investigation and synthetic application were established in the 1960s, primarily through the work of Rolf Huisgen. Hence, the reaction is sometimes referred to as the Huisgen cycloaddition. 1,3-dipolar cycloaddition is an important route to the regio- and stereoselective synthesis of five-membered heterocycles and their ring-opened acyclic derivatives. The dipolarophile is typically an alkene or alkyne, but can be other pi systems. When the dipolarophile is an alkyne, aromatic rings are generally produced.

In organic chemistry, cheletropic reactions, also known as chelotropic reactions, are a type of pericyclic reaction. Specifically, cheletropic reactions are a subclass of cycloadditions. The key distinguishing feature of cheletropic reactions is that on one of the reagents, both new bonds are being made to the same atom.
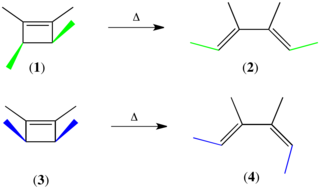
The Woodward–Hoffmann rules are a set of rules devised by Robert Burns Woodward and Roald Hoffmann to rationalize or predict certain aspects of the stereochemistry and activation energy of pericyclic reactions, an important class of reactions in organic chemistry. The rules originate in certain symmetries of the molecule's orbital structure that any molecular Hamiltonian conserves. Consequently, any symmetry-violating reaction must couple extensively to the environment; this imposes an energy barrier on its occurrence, and such reactions are called symmetry-forbidden. Their opposites are symmetry-allowed.
The Hückel method or Hückel molecular orbital theory, proposed by Erich Hückel in 1930, is a simple method for calculating molecular orbitals as linear combinations of atomic orbitals. The theory predicts the molecular orbitals for π-electrons in π-delocalized molecules, such as ethylene, benzene, butadiene, and pyridine. It provides the theoretical basis for Hückel's rule that cyclic, planar molecules or ions with π-electrons are aromatic. It was later extended to conjugated molecules such as pyridine, pyrrole and furan that contain atoms other than carbon and hydrogen (heteroatoms). A more dramatic extension of the method to include σ-electrons, known as the extended Hückel method (EHM), was developed by Roald Hoffmann. The extended Hückel method gives some degree of quantitative accuracy for organic molecules in general and was used to provide computational justification for the Woodward–Hoffmann rules. To distinguish the original approach from Hoffmann's extension, the Hückel method is also known as the simple Hückel method (SHM).

Antarafacial and suprafacial (s) are two topological concepts in organic chemistry describing the relationship between two simultaneous chemical bond making and/or bond breaking processes in or around a reaction center. The reaction center can be a p- or spn-orbital, a conjugated system (π) or even a sigma bond (σ).

In organic chemistry, Möbius aromaticity is a special type of aromaticity believed to exist in a number of organic molecules. In terms of molecular orbital theory these compounds have in common a monocyclic array of molecular orbitals in which there is an odd number of out-of-phase overlaps, the opposite pattern compared to the aromatic character to Hückel systems. The nodal plane of the orbitals, viewed as a ribbon, is a Möbius strip, rather than a cylinder, hence the name. The pattern of orbital energies is given by a rotated Frost circle (with the edge of the polygon on the bottom instead of a vertex), so systems with 4n electrons are aromatic, while those with 4n + 2 electrons are anti-aromatic/non-aromatic. Due to incrementally twisted nature of the orbitals of a Möbius aromatic system, stable Möbius aromatic molecules need to contain at least 8 electrons, although 4 electron Möbius aromatic transition states are well known in the context of the Dewar-Zimmerman framework for pericyclic reactions. Möbius molecular systems were considered in 1964 by Edgar Heilbronner by application of the Hückel method, but the first such isolable compound was not synthesized until 2003 by the group of Rainer Herges. However, the fleeting trans-C9H9+ cation, one conformation of which is shown on the right, was proposed to be a Möbius aromatic reactive intermediate in 1998 based on computational and experimental evidence.
In chemistry, frontier molecular orbital theory is an application of molecular orbital theory describing HOMO–LUMO interactions.
In chemistry, the Möbius–Hückel treatment is a methodology used to predict whether a reaction is allowed or forbidden. It is often used along with the Woodward–Hoffmann approach. The description in this article uses the plus-minus sign notation for parity as shorthand while proceeding around a cycle of orbitals in a molecule or system, while the Woodward–Hoffmann methodology uses a large number of rules with the same consequences.
[6+4] Cycloaddition is a type of cycloaddition between a six-atom pi system and a four-atom pi system, leading to a ten-membered ring. Because this is a higher-order cycloaddition, issues of periselectivity arise in addition to the usual concerns about regio- and stereoselectivity. Six-atom pi systems that have been employed in the reaction include tropone and tropone derivatives, fulvenes, and cycloheptatriene cobalt complexes.
The inverse electron demand Diels–Alder reaction, or DAINV or IEDDA is an organic chemical reaction, in which two new chemical bonds and a six-membered ring are formed. It is related to the Diels–Alder reaction, but unlike the Diels–Alder reaction, the DAINV is a cycloaddition between an electron-rich dienophile and an electron-poor diene. During a DAINV reaction, three pi-bonds are broken, and two sigma bonds and one new pi-bond are formed. A prototypical DAINV reaction is shown on the right.
A metal-centered cycloaddition is a subtype of the more general class of cycloaddition reactions. In such reactions "two or more unsaturated molecules unite directly to form a ring", incorporating a metal bonded to one or more of the molecules. Cycloadditions involving metal centers are a staple of organic and organometallic chemistry, and are involved in many industrially-valuable synthetic processes.
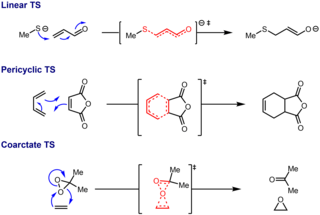
A coarctate reaction is a concerted reaction whose transition state involves two rings, in which at least one atom undergoes the simultaneous making and breaking of two bonds. It is an uncommon reaction topology, compared with linear topology and pericyclic topology. The name is derived from the Latin coarctare, meaning 'to constrict'.