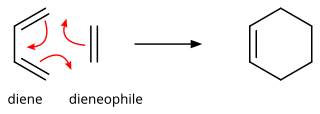
In organic chemistry, the Diels–Alder reaction is a chemical reaction between a conjugated diene and a substituted alkene, commonly termed the dienophile, to form a substituted cyclohexene derivative. It is the prototypical example of a pericyclic reaction with a concerted mechanism. More specifically, it is classified as a thermally allowed [4+2] cycloaddition with Woodward–Hoffmann symbol [π4s + π2s]. It was first described by Otto Diels and Kurt Alder in 1928. For the discovery of this reaction, they were awarded the Nobel Prize in Chemistry in 1950. Through the simultaneous construction of two new carbon–carbon bonds, the Diels–Alder reaction provides a reliable way to form six-membered rings with good control over the regio- and stereochemical outcomes. Consequently, it has served as a powerful and widely applied tool for the introduction of chemical complexity in the synthesis of natural products and new materials. The underlying concept has also been applied to π-systems involving heteroatoms, such as carbonyls and imines, which furnish the corresponding heterocycles; this variant is known as the hetero-Diels–Alder reaction. The reaction has also been generalized to other ring sizes, although none of these generalizations have matched the formation of six-membered rings in terms of scope or versatility. Because of the negative values of ΔH° and ΔS° for a typical Diels–Alder reaction, the microscopic reverse of a Diels–Alder reaction becomes favorable at high temperatures, although this is of synthetic importance for only a limited range of Diels–Alder adducts, generally with some special structural features; this reverse reaction is known as the retro-Diels–Alder reaction.
In chemistry, intramolecular describes a process or characteristic limited within the structure of a single molecule, a property or phenomenon limited to the extent of a single molecule.
In organic chemistry, a cycloaddition is a chemical reaction in which "two or more unsaturated molecules combine with the formation of a cyclic adduct in which there is a net reduction of the bond multiplicity". The resulting reaction is a cyclization reaction. Many but not all cycloadditions are concerted and thus pericyclic. Nonconcerted cycloadditions are not pericyclic. As a class of addition reaction, cycloadditions permit carbon–carbon bond formation without the use of a nucleophile or electrophile.
The 1,3-dipolar cycloaddition is a chemical reaction between a 1,3-dipole and a dipolarophile to form a five-membered ring. The earliest 1,3-dipolar cycloadditions were described in the late 19th century to the early 20th century, following the discovery of 1,3-dipoles. Mechanistic investigation and synthetic application were established in the 1960s, primarily through the work of Rolf Huisgen. Hence, the reaction is sometimes referred to as the Huisgen cycloaddition. 1,3-dipolar cycloaddition is an important route to the regio- and stereoselective synthesis of five-membered heterocycles and their ring-opened acyclic derivatives. The dipolarophile is typically an alkene or alkyne, but can be other pi systems. When the dipolarophile is an alkyne, aromatic rings are generally produced.

The Claisen rearrangement is a powerful carbon–carbon bond-forming chemical reaction discovered by Rainer Ludwig Claisen. The heating of an allyl vinyl ether will initiate a [3,3]-sigmatropic rearrangement to give a γ,δ-unsaturated carbonyl, driven by exergonically favored carbonyl CO bond formation (Δ = −327 kcal/mol.

In organic chemistry, cheletropic reactions, also known as chelotropic reactions, are a type of pericyclic reaction. Specifically, cheletropic reactions are a subclass of cycloadditions. The key distinguishing feature of cheletropic reactions is that on one of the reagents, both new bonds are being made to the same atom.

The Aza-Diels–Alder reaction is a modification of the Diels–Alder reaction wherein a nitrogen replaces sp2 carbon. The nitrogen atom can be part of the diene or the dienophile.
Ring-closing metathesis (RCM) is a widely used variation of olefin metathesis in organic chemistry for the synthesis of various unsaturated rings via the intramolecular metathesis of two terminal alkenes, which forms the cycloalkene as the E- or Z- isomers and volatile ethylene.
The Perkow reaction is an organic reaction in which a trialkyl phosphite ester reacts with a haloketone to form a dialkyl vinyl phosphate and an alkyl halide.

The Wolff rearrangement is a reaction in organic chemistry in which an α-diazocarbonyl compound is converted into a ketene by loss of dinitrogen with accompanying 1,2-rearrangement. The Wolff rearrangement yields a ketene as an intermediate product, which can undergo nucleophilic attack with weakly acidic nucleophiles such as water, alcohols, and amines, to generate carboxylic acid derivatives or undergo [2+2] cycloaddition reactions to form four-membered rings. The mechanism of the Wolff rearrangement has been the subject of debate since its first use. No single mechanism sufficiently describes the reaction, and there are often competing concerted and carbene-mediated pathways; for simplicity, only the textbook, concerted mechanism is shown below. The reaction was discovered by Ludwig Wolff in 1902. The Wolff rearrangement has great synthetic utility due to the accessibility of α-diazocarbonyl compounds, variety of reactions from the ketene intermediate, and stereochemical retention of the migrating group. However, the Wolff rearrangement has limitations due to the highly reactive nature of α-diazocarbonyl compounds, which can undergo a variety of competing reactions.
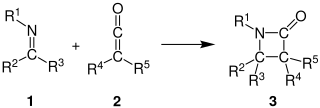
The Staudinger synthesis, also called the Staudinger ketene-imine cycloaddition, is a chemical synthesis in which an imine 1 reacts with a ketene 2 through a non-photochemical 2+2 cycloaddition to produce a β-lactam3. The reaction carries particular importance in the synthesis of β-lactam antibiotics. The Staudinger synthesis should not be confused with the Staudinger reaction, a phosphine or phosphite reaction used to reduce azides to amines.
The divinylcyclopropane-cycloheptadiene rearrangement is an organic chemical transformation that involves the isomerization of a 1,2-divinylcyclopropane into a cycloheptadiene or -triene. It is conceptually related to the Cope rearrangement, but has the advantage of a strong thermodynamic driving force due to the release of ring strain. This thermodynamic power is recently being considered as an alternative energy source.
Trimethylenemethane cycloaddition is the formal (3+2) annulation of trimethylenemethane (TMM) derivatives to two-atom pi systems. Although TMM itself is too reactive and unstable to be stored, reagents which can generate TMM or TMM synthons in situ can be used to effect cycloaddition reactions with appropriate electron acceptors. Generally, electron-deficient pi bonds undergo cyclization with TMMs more easily than electron-rich pi bonds.
A (4+3) cycloaddition is a cycloaddition between a four-atom π-system and a three-atom π-system to form a seven-membered ring. Allyl or oxyallyl cations (propenylium-2-olate) are commonly used three-atom π-systems, while a diene plays the role of the four-atom π-system. It represents one of the relatively few synthetic methods available to form seven-membered rings stereoselectively in high yield.
[6+4] Cycloaddition is a type of cycloaddition between a six-atom pi system and a four-atom pi system, leading to a ten-membered ring. Because this is a higher-order cycloaddition, issues of periselectivity arise in addition to the usual concerns about regio- and stereoselectivity. Six-atom pi systems that have been employed in the reaction include tropone and tropone derivatives, fulvenes, and cycloheptatriene cobalt complexes.
In organic chemistry, enone–alkene cycloadditions are a version of the [2+2] cycloaddition. This reaction involves an enone and alkene as substrates. Although the concerted photochemical [2+2] cycloaddition is allowed, the reaction between enones and alkenes is stepwise and involves discrete diradical intermediates.
Phenol oxidation with hypervalent iodine reagents leads to the formation of quinone-type products or iodonium ylides, depending on the structure of the phenol. Trapping of either product is possible with a suitable reagent, and this method is often employed in tandem with a second process.
The inverse electron demand Diels–Alder reaction, or DAINV or IEDDA is an organic chemical reaction, in which two new chemical bonds and a six-membered ring are formed. It is related to the Diels–Alder reaction, but unlike the Diels–Alder reaction, the DAINV is a cycloaddition between an electron-rich dienophile and an electron-poor diene. During a DAINV reaction, three pi-bonds are broken, and two sigma bonds and one new pi-bond are formed. A prototypical DAINV reaction is shown on the right.
A metal-centered cycloaddition is a subtype of the more general class of cycloaddition reactions. In such reactions "two or more unsaturated molecules unite directly to form a ring", incorporating a metal bonded to one or more of the molecules. Cycloadditions involving metal centers are a staple of organic and organometallic chemistry, and are involved in many industrially-valuable synthetic processes.
The Danheiser benzannulation is a chemical reaction used in organic chemistry to generate highly substituted phenols in a single step. It is named after Rick L. Danheiser who developed the reaction.










