Sharpless asymmetric dihydroxylation is the chemical reaction of an alkene with osmium tetroxide in the presence of a chiral quinine ligand to form a vicinal diol. The reaction has been applied to alkenes of virtually every substitution, often high enantioselectivities are realized, with the chiral outcome controlled by the choice of dihydroquinidine (DHQD) vs dihydroquinine (DHQ) as the ligand. Asymmetric dihydroxylation reactions are also highly site selective, providing products derived from reaction of the most electron-rich double bond in the substrate.
In chemistry, an electrophile is a chemical species that forms bonds with nucleophiles by accepting an electron pair. Because electrophiles accept electrons, they are Lewis acids. Most electrophiles are positively charged, have an atom that carries a partial positive charge, or have an atom that does not have an octet of electrons.
The Heck reaction is the chemical reaction of an unsaturated halide with an alkene in the presence of a base and a palladium catalyst to form a substituted alkene. It is named after Tsutomu Mizoroki and Richard F. Heck. Heck was awarded the 2010 Nobel Prize in Chemistry, which he shared with Ei-ichi Negishi and Akira Suzuki, for the discovery and development of this reaction. This reaction was the first example of a carbon-carbon bond-forming reaction that followed a Pd(0)/Pd(II) catalytic cycle, the same catalytic cycle that is seen in other Pd(0)-catalyzed cross-coupling reactions. The Heck reaction is a way to substitute alkenes.
The Stille reaction is a chemical reaction widely used in organic synthesis. The reaction involves the coupling of two organic groups, one of which is carried as an organotin compound (also known as organostannanes). A variety of organic electrophiles provide the other coupling partner. The Stille reaction is one of many palladium-catalyzed coupling reactions.
The Suzuki reaction or Suzuki coupling is an organic reaction that uses a palladium complex catalyst to cross-couple a boronic acid to an organohalide. It was first published in 1979 by Akira Suzuki, and he shared the 2010 Nobel Prize in Chemistry with Richard F. Heck and Ei-ichi Negishi for their contribution to the discovery and development of noble metal catalysis in organic synthesis. This reaction is sometimes telescoped with the related Miyaura borylation; the combination is the Suzuki–Miyaura reaction. It is widely used to synthesize polyolefins, styrenes, and substituted biphenyls.

Organoboron chemistry or organoborane chemistry studies organoboron compounds, also called organoboranes. These chemical compounds combine boron and carbon; typically, they are organic derivatives of borane (BH3), as in the trialkyl boranes.

The Bamford–Stevens reaction is a chemical reaction whereby treatment of tosylhydrazones with strong base gives alkenes. It is named for the British chemist William Randall Bamford and the Scottish chemist Thomas Stevens Stevens (1900–2000). The usage of aprotic solvents gives predominantly Z-alkenes, while protic solvent gives a mixture of E- and Z-alkenes. As an alkene-generating transformation, the Bamford–Stevens reaction has broad utility in synthetic methodology and complex molecule synthesis.
Organoselenium chemistry is the science exploring the properties and reactivity of organoselenium compounds, chemical compounds containing carbon-to-selenium chemical bonds. Selenium belongs with oxygen and sulfur to the group 16 elements or chalcogens, and similarities in chemistry are to be expected. Organoselenium compounds are found at trace levels in ambient waters, soils and sediments.

The Peterson olefination is the chemical reaction of α-silyl carbanions with ketones to form a β-hydroxysilane (2) which eliminates to form alkenes (3).

Organozinc chemistry is the study of the physical properties, synthesis, and reactions of organozinc compounds, which are organometallic compounds that contain carbon (C) to zinc (Zn) chemical bonds.
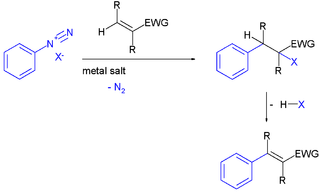
The Meerwein arylation is an organic reaction involving the addition of an aryl diazonium salt (ArN2X) to an electron-poor alkene usually supported by a metal salt. The reaction product is an alkylated arene compound. The reaction is named after Hans Meerwein, one of its inventors who first published it in 1939.
In organic chemistry, the Ei mechanism, also known as a thermal syn elimination or a pericyclic syn elimination, is a special type of elimination reaction in which two vicinal (adjacent) substituents on an alkane framework leave simultaneously via a cyclic transition state to form an alkene in a syn elimination. This type of elimination is unique because it is thermally activated and does not require additional reagents, unlike regular eliminations, which require an acid or base, or would in many cases involve charged intermediates. This reaction mechanism is often found in pyrolysis.
Organotellurium chemistry describes the synthesis and properties of organotellurium compounds, chemical compounds containing a carbon-tellurium chemical bond. Organotellurium chemistry is a lightly studied area, in part because of it having few applications.
Selenoxide elimination is a method for the chemical synthesis of alkenes from selenoxides. It is most commonly used to synthesize α,β-unsaturated carbonyl compounds from the corresponding saturated analogues. It is mechanistically related to the Cope reaction.
The nitrone-olefin (3+2) cycloaddition reaction is the combination of a nitrone with an alkene or alkyne to generate an isoxazoline or isoxazolidine via a (3+2) cycloaddition process. This reaction is a 1,3-dipolar cycloaddition, in which the nitrone acts as the 1,3-dipole, and the alkene or alkyne as the dipolarophile.

Reductions with samarium(II) iodide involve the conversion of various classes of organic compounds into reduced products through the action of samarium(II) iodide, a mild one-electron reducing agent.
Desulfonylation reactions are chemical reactions leading to the removal of a sulfonyl group from organic compounds. As the sulfonyl functional group is electron-withdrawing, methods for cleaving the sulfur–carbon bonds of sulfones are typically reductive in nature. Olefination or replacement with hydrogen may be accomplished using reductive desulfonylation methods.
In organic chemistry, the Baylis–Hillman, Morita–Baylis–Hillman, or MBH reaction is a carbon-carbon bond-forming reaction between an activated alkene and a carbon electrophile in the presence of a nucleophilic catalyst, such as a tertiary amine or phosphine. The product is densely functionalized, joining the alkene at the α-position to a reduced form of the electrophile.

Trifluoroperacetic acid is an organofluorine compound, the peroxy acid analog of trifluoroacetic acid, with the condensed structural formula CF
3COOOH. It is a strong oxidizing agent for organic oxidation reactions, such as in Baeyer–Villiger oxidations of ketones. It is the most reactive of the organic peroxy acids, allowing it to successfully oxidise relatively unreactive alkenes to epoxides where other peroxy acids are ineffective. It can also oxidise the chalcogens in some functional groups, such as by transforming selenoethers to selones. It is a potentially explosive material and is not commercially available, but it can be quickly prepared as needed. Its use as a laboratory reagent was pioneered and developed by William D. Emmons.
In organic chemistry, hydrovinylation is the formal insertion of an alkene into the C-H bond of ethylene :
