In chemistry, an electrophile is a chemical species that forms bonds with nucleophiles by accepting an electron pair. Because electrophiles accept electrons, they are Lewis acids. Most electrophiles are positively charged, have an atom that carries a partial positive charge, or have an atom that does not have an octet of electrons.

An enamine is an unsaturated compound derived by the condensation of an aldehyde or ketone with a secondary amine. Enamines are versatile intermediates.
The Wolff–Kishner reduction is a reaction used in organic chemistry to convert carbonyl functionalities into methylene groups. In the context of complex molecule synthesis, it is most frequently employed to remove a carbonyl group after it has served its synthetic purpose of activating an intermediate in a preceding step. As such, there is no obvious retron for this reaction. The reaction was reported by Nikolai Kischner in 1911 and Ludwig Wolff in 1912.
The Heck reaction is the chemical reaction of an unsaturated halide with an alkene in the presence of a base and a palladium catalyst to form a substituted alkene. It is named after Tsutomu Mizoroki and Richard F. Heck. Heck was awarded the 2010 Nobel Prize in Chemistry, which he shared with Ei-ichi Negishi and Akira Suzuki, for the discovery and development of this reaction. This reaction was the first example of a carbon-carbon bond-forming reaction that followed a Pd(0)/Pd(II) catalytic cycle, the same catalytic cycle that is seen in other Pd(0)-catalyzed cross-coupling reactions. The Heck reaction is a way to substitute alkenes.
In organic chemistry, the diazo group is an organic moiety consisting of two linked nitrogen atoms at the terminal position. Overall charge-neutral organic compounds containing the diazo group bound to a carbon atom are called diazo compounds or diazoalkanes and are described by the general structural formula R2C=N+=N−. The simplest example of a diazo compound is diazomethane, CH2N2. Diazo compounds should not be confused with azo compounds or with diazonium compounds.

The Bamford–Stevens reaction is a chemical reaction whereby treatment of tosylhydrazones with strong base gives alkenes. It is named for the British chemist William Randall Bamford and the Scottish chemist Thomas Stevens Stevens (1900–2000). The usage of aprotic solvents gives predominantly Z-alkenes, while protic solvent gives a mixture of E- and Z-alkenes. As an alkene-generating transformation, the Bamford–Stevens reaction has broad utility in synthetic methodology and complex molecule synthesis.
Dichlorocarbene is the reactive intermediate with chemical formula CCl2. Although this chemical species has not been isolated, it is a common intermediate in organic chemistry, being generated from chloroform. This bent diamagnetic molecule rapidly inserts into other bonds.
The Shapiro reaction or tosylhydrazone decomposition is an organic reaction in which a ketone or aldehyde is converted to an alkene through an intermediate hydrazone in the presence of 2 equivalents of organolithium reagent. The reaction was discovered by Robert H. Shapiro in 1967. The Shapiro reaction was used in the Nicolaou Taxol total synthesis. This reaction is very similar to the Bamford–Stevens reaction, which also involves the basic decomposition of tosyl hydrazones.

The Danishefsky Taxol total synthesis in organic chemistry is an important third Taxol synthesis published by the group of Samuel Danishefsky in 1996 two years after the first two efforts described in the Holton Taxol total synthesis and the Nicolaou Taxol total synthesis. Combined they provide a good insight in the application of organic chemistry in total synthesis.
The Barton–Kellogg reaction is a coupling reaction between a diazo compound and a thioketone, giving an alkene by way of an episulfide intermediate. The Barton–Kellogg reaction is also known as Barton–Kellogg olefination and Barton olefin synthesis.
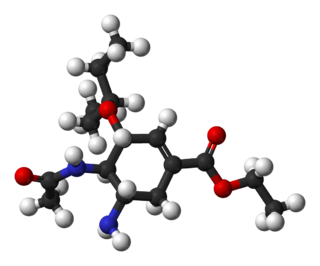
Oseltamivir total synthesis concerns the total synthesis of the antiinfluenza drug oseltamivir marketed by Hoffmann-La Roche under the trade name Tamiflu. Its commercial production starts from the biomolecule shikimic acid harvested from Chinese star anise and from recombinant E. coli. Control of stereochemistry is important: the molecule has three stereocenters and the sought-after isomer is only 1 of 8 stereoisomers.
The Barton reaction, also known as the Barton nitrite ester reaction, is a photochemical reaction that involves the photolysis of an alkyl nitrite to form a δ-nitroso alcohol.
In organic chemistry, the Ei mechanism, also known as a thermal syn elimination or a pericyclic syn elimination, is a special type of elimination reaction in which two vicinal (adjacent) substituents on an alkane framework leave simultaneously via a cyclic transition state to form an alkene in a syn elimination. This type of elimination is unique because it is thermally activated and does not require additional reagents, unlike regular eliminations, which require an acid or base, or would in many cases involve charged intermediates. This reaction mechanism is often found in pyrolysis.
The Hofmann–Löffler reaction (also referred to as Hofmann–Löffler–Freytag reaction, Löffler–Freytag reaction, Löffler–Hofmann reaction, as well as Löffler's method) is an organic reaction in which a cyclic amine 2 (pyrrolidine or, in some cases, piperidine) is generated by thermal or photochemical decomposition of N-halogenated amine 1 in the presence of a strong acid (concentrated sulfuric acid or concentrated CF3CO2H). The Hofmann–Löffler–Freytag reaction proceeds via an intramolecular hydrogen atom transfer to a nitrogen-centered radical and is an example of a remote intramolecular free radical C–H functionalization.
In organic chemistry, α-halo ketones can be reduced with loss of the halogen atom to form enolates. The α-halo ketones are readily prepared from ketones by various ketone halogenation reactions, and the products are reactive intermediates that can be used for a variety of other chemical reactions.
Metal-catalyzed cyclopropanations are chemical reactions that result in the formation of a cyclopropane ring from a metal carbenoid species and an alkene. In the Simmons–Smith reaction the metal involved is zinc. Metal carbenoid species can be generated through the reaction of a diazo compound with a transition metal). The intramolecular variant of this reaction was first reported in 1961. Rhodium carboxylate complexes, such as dirhodium tetraacetate, are common catalysts. Enantioselective cyclopropanations have been developed.
An insertion reaction is a chemical reaction where one chemical entity interposes itself into an existing bond of typically a second chemical entity e.g.:
In organic chemistry, the Baylis–Hillman, Morita–Baylis–Hillman, or MBH reaction is a carbon-carbon bond-forming reaction between an activated alkene and a carbon electrophile in the presence of a nucleophilic catalyst, such as a tertiary amine or phosphine. The product is densely functionalized, joining the alkene at the α-position to a reduced form of the electrophile.

Tetraethylammonium iodide is a quaternary ammonium compound with the chemical formula C8H20N+I−. It has been used as the source of tetraethylammonium ions in pharmacological and physiological studies, but is also used in organic chemical synthesis.
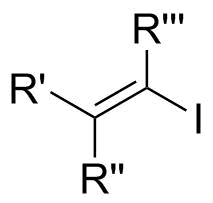
In organic chemistry, a vinyl iodide functional group is an alkene with one or more iodide substituents. Vinyl iodides are versatile molecules that serve as important building blocks and precursors in organic synthesis. They are commonly used in carbon-carbon forming reactions in transition-metal catalyzed cross-coupling reactions, such as Stille reaction, Heck reaction, Sonogashira coupling, and Suzuki coupling. Synthesis of well-defined geometry or complexity vinyl iodide is important in stereoselective synthesis of natural products and drugs.




