In chemistry, an alkene is a hydrocarbon that contains a carbon–carbon double bond.
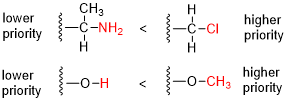
The Cahn–Ingold–Prelog (CIP) sequence rules, named for organic chemists Robert Sidney Cahn, Christopher Kelk Ingold, and Vladimir Prelog — alternatively termed the CIP priority rules, system, or conventions — are a standard process used in organic chemistry to completely and unequivocally name a stereoisomer of a molecule. The purpose of the CIP system is to assign an R or S descriptor to each stereocenter and an E or Z descriptor to each double bond so that the configuration of the entire molecule can be specified uniquely by including the descriptors in its systematic name. A molecule may contain any number of stereocenters and any number of double bonds, and each usually gives rise to two possible isomers. A molecule with an integer n describing the number of its stereogenic centers will usually have 2n stereoisomers, and 2n−1 diastereomers each having an associated pair of enantiomers. The CIP sequence rules contribute to the precise naming of every stereoisomer of every organic and organometallic molecule with all atoms of ligancy of fewer than 4.
In organic chemistry, the Diels–Alder reaction is a chemical reaction between a conjugated diene and a substituted alkene, commonly termed the dienophile, to form a substituted cyclohexene derivative. It is the prototypical example of a pericyclic reaction with a concerted mechanism. More specifically, it is classified as a thermally-allowed [4+2] cycloaddition with Woodward–Hoffmann symbol [π4s + π2s]. It was first described by Otto Diels and Kurt Alder in 1928. For the discovery of this reaction, they were awarded the Nobel Prize in Chemistry in 1950. Through the simultaneous construction of two new carbon–carbon bonds, the Diels–Alder reaction provides a reliable way to form six-membered rings with good control over the regio- and stereochemical outcomes. Consequently, it has served as a powerful and widely applied tool for the introduction of chemical complexity in the synthesis of natural products and new materials. The underlying concept has also been applied to π-systems involving heteroatoms, such as carbonyls and imines, which furnish the corresponding heterocycles; this variant is known as the hetero-Diels–Alder reaction. The reaction has also been generalized to other ring sizes, although none of these generalizations have matched the formation of six-membered rings in terms of scope or versatility. Because of the negative values of ΔH° and ΔS° for a typical Diels–Alder reaction, the microscopic reverse of a Diels–Alder reaction becomes favorable at high temperatures, although this is of synthetic importance for only a limited range of Diels-Alder adducts, generally with some special structural features; this reverse reaction is known as the retro-Diels–Alder reaction.
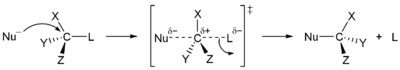
In organic chemistry and inorganic chemistry, nucleophilic substitution is a fundamental class of reactions in which a leaving group is replaced by an electron rich compound (nucleophile). The whole molecular entity of which the electrophile and the leaving group are part is usually called the substrate. The nucleophile essentially attempts to replace the leaving group as the primary substituent in the reaction itself, as a part of another molecule.
The SN1 reaction is a substitution reaction in organic chemistry, the name of which refers to the Hughes-Ingold symbol of the mechanism. "SN" stands for "nucleophilic substitution", and the "1" says that the rate-determining step is unimolecular. Thus, the rate equation is often shown as having first-order dependence on electrophile and zero-order dependence on nucleophile. This relationship holds for situations where the amount of nucleophile is much greater than that of the intermediate. Instead, the rate equation may be more accurately described using steady-state kinetics. The reaction involves a carbocation intermediate and is commonly seen in reactions of secondary or tertiary alkyl halides under strongly basic conditions or, under strongly acidic conditions, with secondary or tertiary alcohols. With primary and secondary alkyl halides, the alternative SN2 reaction occurs. In inorganic chemistry, the SN1 reaction is often known as the dissociative mechanism. This dissociation pathway is well-described by the cis effect. A reaction mechanism was first proposed by Christopher Ingold et al. in 1940. This reaction does not depend much on the strength of the nucleophile unlike the SN2 mechanism. This type of mechanism involves two steps. The first step is the reversible ionization of alkyl halide in the presence of aqueous acetone or ethyl alcohol. This step provides a carbocation as an intermediate.
A halogen addition reaction is a simple organic reaction where a halogen molecule is added to the carbon–carbon double bond of an alkene functional group.
The oxymercuration reaction is an electrophilic addition organic reaction that transforms an alkene into a neutral alcohol. In oxymercuration, the alkene reacts with mercuric acetate (AcO–Hg–OAc) in aqueous solution to yield the addition of an acetoxymercury (HgOAc) group and a hydroxy (OH) group across the double bond. Carbocations are not formed in this process and thus rearrangements are not observed. The reaction follows Markovnikov's rule and it is an anti addition.

The aldol reaction is a means of forming carbon–carbon bonds in organic chemistry. Discovered independently by the Russian chemist Alexander Borodin in 1869 and by the French chemist Charles-Adolphe Wurtz in 1872, the reaction combines two carbonyl compounds to form a new β-hydroxy carbonyl compound. These products are known as aldols, from the aldehyde + alcohol, a structural motif seen in many of the products. Aldol structural units are found in many important molecules, whether naturally occurring or synthetic. For example, the aldol reaction has been used in the large-scale production of the commodity chemical pentaerythritol and the synthesis of the heart disease drug Lipitor.
Isomerases are a general class of enzymes that convert a molecule from one isomer to another. Isomerases facilitate intramolecular rearrangements in which bonds are broken and formed. The general form of such a reaction is as follows:
The 1,3-dipolar cycloaddition is a chemical reaction between a 1,3-dipole and a dipolarophile to form a five-membered ring. The earliest 1,3-dipolar cycloadditions were described in the late 19th century to the early 20th century, following the discovery of 1,3-dipoles. Mechanistic investigation and synthetic application were established in the 1960s, primarily through the work of Rolf Huisgen. Hence, the reaction is sometimes referred to as the Huisgen cycloaddition. 1,3-dipolar cycloaddition is an important route to the regio- and stereoselective synthesis of five-membered heterocycles and their ring-opened acyclic derivatives. The dipolarophile is typically an alkene or alkyne, but can be other pi systems. When the dipolarophile is an alkyne, aromatic rings are generally produced.
In chemistry, stereoselectivity is the property of a chemical reaction in which a single reactant forms an unequal mixture of stereoisomers during a non-stereospecific creation of a new stereocenter or during a non-stereospecific transformation of a pre-existing one. The selectivity arises from differences in steric effects and electronic effects in the mechanistic pathways leading to the different products. Stereoselectivity can vary in degree but it can never be total since the activation energy difference between the two pathways is finite. Both products are at least possible and merely differ in amount. However, in favorable cases, the minor stereoisomer may not be detectable by the analytic methods used.
An allylic rearrangement or allylic shift is an organic reaction in which the double bond in an allyl chemical compound shifts to the next carbon atom. It is encountered in nucleophilic substitution.
The Wittig reaction or Wittig olefination is a chemical reaction of an aldehyde or ketone with a triphenyl phosphonium ylide to give an alkene and triphenylphosphine oxide.
Neighbouring group participation (NGP) in organic chemistry has been defined by IUPAC as the interaction of a reaction centre with a lone pair of electrons in an atom or the electrons present in a sigma bond or pi bond contained within the parent molecule but not conjugated with the reaction centre. When NGP is in operation it is normal for the reaction rate to be increased. It is also possible for the stereochemistry of the reaction to be abnormal when compared with a normal reaction. While it is possible for neighbouring groups to influence many reactions in organic chemistry this page is limited to neighbouring group effects seen with carbocations and SN2 reactions.
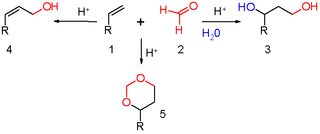
The Prins reaction is an organic reaction consisting of an electrophilic addition of an aldehyde or ketone to an alkene or alkyne followed by capture of a nucleophile or elimination of an H+ ion. The outcome of the reaction depends on reaction conditions. With water and a protic acid such as sulfuric acid as the reaction medium and formaldehyde the reaction product is a 1,3-diol. When water is absent, the cationic intermediate loses a proton to give an allylic alcohol. With an excess of formaldehyde and a low reaction temperature the reaction product is a dioxane. When water is replaced by acetic acid the corresponding esters are formed.

The Darzens reaction is the chemical reaction of a ketone or aldehyde with an α-haloester in the presence of a base to form an α,β-epoxy ester, also called a "glycidic ester". This reaction was discovered by the organic chemist Auguste Georges Darzens in 1904.

In stereochemistry, asymmetric induction describes the preferential formation in a chemical reaction of one enantiomer or diastereoisomer over the other as a result of the influence of a chiral feature present in the substrate, reagent, catalyst or environment. Asymmetric induction is a key element in asymmetric synthesis.

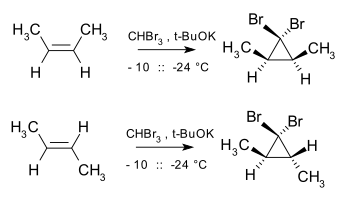
Cyclopropanation refers to any chemical process which generates cyclopropane rings. It is an important process in modern chemistry as many useful compounds bear this motif; for example pyrethroids and a number of quinolone antibiotics. However the high ring strain present in cyclopropanes makes them challenging to produce and generally requires the use of highly reactive species, such as carbenes, ylids and carbanions. Many of the reactions proceed in a cheletropic manner.
The Fürst-Plattner rule describes the stereoselective addition of nucleophiles to cyclohexene derivatives.
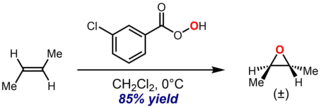
The Prilezhaev reaction, also known as the Prileschajew reaction or Prilezhaev epoxidation, is the chemical reaction of an alkene with a peroxy acid to form epoxides. It is named after Nikolai Prilezhaev, who first reported this reaction in 1909. A widely used peroxy acid for this reaction is meta-chloroperoxybenzoic acid (m-CPBA), due to its stability and good solubility in most organic solvents. An illustrative example is the epoxidation of trans-2-butene with m-CPBA to give trans-2,3-epoxybutane: