The following outline is provided as an overview of and topical guide to organic chemistry:

Diazomethane is the chemical compound CH2N2, discovered by German chemist Hans von Pechmann in 1894. It is the simplest diazo compound. In the pure form at room temperature, it is an extremely sensitive explosive yellow gas; thus, it is almost universally used as a solution in diethyl ether. The compound is a popular methylating agent in the laboratory, but it is too hazardous to be employed on an industrial scale without special precautions. Use of diazomethane has been significantly reduced by the introduction of the safer and equivalent reagent trimethylsilyldiazomethane.
The diazogroup is an organic moiety consisting of two linked nitrogen atoms (azo) at the terminal position. Overall charge neutral organic compounds containing the diazo group bound to a carbon atom are called diazo compounds or diazoalkanes and are described by the general structural formula R2C=N+=N–. The simplest example of a diazo compound is diazomethane, CH2N2. Diazo compounds (R2C=N2) should not be confused with azo compounds of the type R-N=N-R or with diazonium compounds of the type R-N2+.
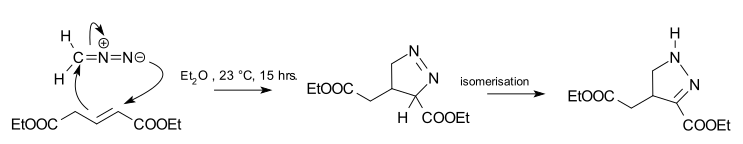
The 1,3-dipolar cycloaddition is a chemical reaction between a 1,3-dipole and a dipolarophile to form a five-membered ring. The earliest 1,3-dipolar cycloadditions were described in the late 19th century to the early 20th century, following the discovery of 1,3-dipoles. Mechanistic investigation and synthetic application were established in the 1960s, primarily through the work of Rolf Huisgen. Hence, the reaction is sometimes referred to as the Huisgen cycloaddition. 1,3-dipolar cycloaddition is an important route to the regio- and stereoselective synthesis of five-membered heterocycles and their ring-opened acyclic derivatives. The dipolarophile is typically an alkene or alkyne, but can be other pi systems. When the dipolarophile is an alkyne, aromatic rings are generally produced.

On water reactions are a group of organic reactions that take place as an emulsion in water and that exhibit an unusual reaction rate acceleration compared to the same reaction in an organic solvent or compared to the corresponding dry media reaction. This effect has been known for many years but in 2005 researchers in the group of K. Barry Sharpless presented a systematic study into this phenomenon.
In organic chemistry, the Arndt–Eistert reaction is the conversion of a carboxylic acid to its homologue. Named for the German chemists Fritz Arndt (1885–1969) and Bernd Eistert (1902–1978), the method entails treating an acid chlorides with diazomethane. It is a popular method of producing β-amino acids from α-amino acids.

The Povarov reaction is an organic reaction described as a formal cycloaddition between an aromatic imine and an alkene. The imine in this organic reaction is a condensation reaction product from an aniline type compound and a benzaldehyde type compound. The alkene must be electron rich which means that functional groups attached to the alkene must be able to donate electrons. Such alkenes are enol ethers and enamines. The reaction product in the original Povarov reaction is a quinoline. Because the reactions can be carried out with the three components premixed in one reactor it is an example of a multi-component reaction.

Trimethylsilyldiazomethane is the organosilicon compound with the formula (CH3)3SiCHN2. It is classified as a diazo compound. Trimethylsilyldiazomethane is a commercially available reagent used in organic chemistry as a methylating agent and as a source of CH2 group. Its behavior is akin to the less convenient reagent diazomethane.

Azomethine ylides are nitrogen-based 1,3-dipoles, consisting of an iminium ion next to a carbanion. They are used in 1,3-dipolar cycloaddition reactions to form five-membered heterocycles, including pyrrolidines and pyrrolines. These reactions are highly stereo- and regioselective, and have the potential to form four new contiguous stereocenters. Azomethine ylides thus have high utility in total synthesis, and formation of chiral ligands and pharmaceuticals. Azomethine ylides can be generated from many sources, including aziridines, imines, and iminiums. They are often generated in situ, and immediately reacted with dipolarophiles.

The Prato reaction is a particular example of the well-known 1,3-dipolar cycloaddition of azomethine ylides to olefins. In fullerene chemistry this reaction refers to the functionalization of fullerenes and nanotubes. The amino acid sarcosine reacts with paraformaldehyde when heated at reflux in toluene to an ylide which reacts with a double bond in a 6,6 ring position in a fullerene via a 1,3-dipolar cycloaddition to yield a N-methylpyrrolidine derivative or pyrrolidinofullerene or pyrrolidino[[3,4:1,2]] [60]fullerene in 82% yield based on C60 conversion.

Hydrazone iodination is an organic reaction in which a hydrazone is converted into a vinyl iodide by reaction of iodine and a non-nucleophilic base such as DBU. First published by Derek Barton in 1962 the reaction is sometimes referred to as the Barton reaction or, more descriptively, as the Barton vinyl iodine procedure.
A nitrone is a functional group in organic chemistry consisting of an N-oxide of an imine. The general structure is R1R2C=NR3+O− where R3 is not H. A nitrone is a 1,3-dipole, and is used in 1,3-dipolar cycloadditions. Other reactions of nitrones are known, including formal [3+3] cycloadditions to form 6-membered rings, as well as formal [5+2] cycloadditions to form 7-membered rings. Nitrones should not be confused with nitrenes.
The Demjanov rearrangement is the chemical reaction of primary amines with nitrous acid to give rearranged alcohols. It involves substitution by a hydroxyl group with a possible ring expansion. It is named after the Russian chemist Nikolai Jakovlevich Demjanov (1861–1938).

The Wolff rearrangement is a reaction in organic chemistry in which an α-diazocarbonyl compound is converted into a ketene by loss of dinitrogen with accompanying 1,2-rearrangement. The Wolff rearrangement yields a ketene as an intermediate product, which can undergo nucleophilic attack with weakly acidic nucleophiles such as water, alcohols, and amines, to generate carboxylic acid derivatives or undergo [2+2] cycloaddition reactions to form four-membered rings. The mechanism of the Wolff rearrangement has been the subject of debate since its first use. No single mechanism sufficiently describes the reaction, and there are often competing concerted and carbene-mediated pathways; for simplicity, only the textbook, concerted mechanism is shown below. The reaction was discovered by Ludwig Wolff in 1902. The Wolff rearrangement has great synthetic utility due to the accessibility of α-diazocarbonyl compounds, variety of reactions from the ketene intermediate, and stereochemical retention of the migrating group. However, the Wolff rearrangement has limitations due to the highly reactive nature of α-diazocarbonyl compounds, which can undergo a variety of competing reactions.

Cyclopropanation refers to any chemical process which generates cyclopropane rings. It is an important process in modern chemistry as many useful compounds bear this motif; for example pyrethroids and a number of quinolone antibiotics. However the high ring strain present in cyclopropanes makes them challenging to produce and generally requires the use of highly reactive species, such as carbenes, ylids and carbanions. Many of the reactions proceed in a cheletropic manner.
The nitrone-olefin (3+2) cycloaddition reaction is the combination of a nitrone with an alkene or alkyne to generate an isoxazoline or isoxazolidine via a [3+2] cycloaddition process. This reaction is a 1,3-dipolar cycloaddition, in which the nitrone acts as the 1,3-dipole, and the alkene or alkyne as the dipolarophile.
In organic chemistry, a dipolar compound or simply dipole is an electrically neutral molecule carrying a positive and a negative charge in at least one canonical description. In most dipolar compounds the charges are delocalized. Unlike salts, dipolar compounds have charges on separate atoms, not on positive and negative ions that make up the compound. Dipolar compounds exhibit a dipole moment.
The term bioorthogonal chemistry refers to any chemical reaction that can occur inside of living systems without interfering with native biochemical processes. The term was coined by Carolyn R. Bertozzi in 2003. Since its introduction, the concept of the bioorthogonal reaction has enabled the study of biomolecules such as glycans, proteins, and lipids in real time in living systems without cellular toxicity. A number of chemical ligation strategies have been developed that fulfill the requirements of bioorthogonality, including the 1,3-dipolar cycloaddition between azides and cyclooctynes, between nitrones and cyclooctynes, oxime/hydrazone formation from aldehydes and ketones, the tetrazine ligation, the isocyanide-based click reaction, and most recently, the quadricyclane ligation.
The Buchner–Curtius–Schlotterbeck reaction is the reaction of aldehydes or ketones with aliphatic diazoalkanes to form homologated ketones. It was first described by Eduard Buchner and Theodor Curtius in 1885 and later by Fritz Schlotterbeck in 1907. Two German chemists also preceded Schlotterbeck in discovery of the reaction, Hans von Pechmann in 1895 and Viktor Meyer in 1905. The reaction has since been extended to the synthesis of β-keto esters from the condensation between aldehydes and diazo esters. The general reaction scheme is as follows:
The Danheiser benzannulation is a chemical reaction used in organic chemistry to generate highly substituted phenols in a single step. It is named after Rick Danheiser who developed the reaction.