System of connected p-orbitals with delocalized electrons in a molecule
Cinnamaldehyde is a naturally-occurring compound that has a conjugated systempenta-1,3-diene is a molecule with a conjugated systemDiazomethane has a conjugated pi-system
A conjugated system has a region of overlapping p-orbitals, bridging the interjacent locations that simple diagrams illustrate as not having a π bond. They allow a delocalization of π electrons across all the adjacent aligned p-orbitals.[3] The π electrons do not belong to a single bond or atom, but rather to a group of atoms.
Molecules containing conjugated systems of orbitals and electrons are called conjugated molecules, which have overlapping p orbitals on three or more atoms. Some simple organic conjugated molecules are 1,3-butadiene, benzene, and allylic carbocations.[4] The largest conjugated systems are found in graphene, graphite, conductive polymers and carbon nanotubes.
Chemical bonding in conjugated systems
Some prototypical examples of species with delocalized bonding. Top row: pyridine, furan, tropylium cation. Second row: allyl radical, acetate ion, acrolein. Atoms involved are in bold red, while electrons involved in delocalized bonding are in blue. (Particular attention should be paid to the involvement or non-involvement of "non-bonding" electrons.)
Conjugation is possible by means of alternating single and double bonds in which each atom supplies a p orbital perpendicular to the plane of the molecule. However, that is not the only way for conjugation to take place. As long as each contiguous atom in a chain has an available p orbital, the system can be considered conjugated. For example, furan is a five-membered ring with two alternating double bonds flanking an oxygen.[5] The oxygen has two lone pairs, one of which occupies a p orbital perpendicular to the ring on that position, thereby maintaining the conjugation of that five-membered ring by overlap with the perpendicular p orbital on each of the adjacent carbon atoms. The other lone pair remains in plane and does not participate in conjugation.
In general, any sp2 or sp-hybridized carbon or heteroatom, including ones bearing an empty orbital or lone pair orbital, can participate in conjugated systems. However lone pairs do not always participate in a conjugated system. For example, in pyridine, the nitrogen atom already participates in the conjugated system through a formal double bond with an adjacent carbon, so the lone pair remains in the plane of the ring in an sp2 hybrid orbital and does not participate in the conjugation. A requirement for conjugation is orbital overlap. Thus, the conjugated system must be planar (or nearly so). As a consequence, lone pairs which do participate in conjugated systems will occupy orbitals of pure p character instead of spn hybrid orbitals typical for nonconjugated lone pairs.
The π system of furan and lone pairs. Note that one of the oxygen lone pairs participates in conjugation in a p orbital, while the other lone pair is in an sp hybridized orbital in the plane of the molecule and not part of the π system. The participation of six electrons in the π system makes furan aromatic (see below).
A common model for the treatment of conjugated molecules is a composite valence bond / Hückel molecular orbital theory (VB/HMOT) treatment, in which the σ framework of the molecule is separated from the π system (or systems) of the molecule (see the article on the sigma-pi and equivalent-orbital models for this model and an alternative treatment). Although σ bonding can be treated using a delocalized approach as well, it is generally the π bonding that is being considered when delocalized bonding is invoked in the context of simple organic molecules.
Sigma (σ) framework: The σ framework is described by a strictly localized bonding scheme and consists of σ bonds formed from the interactions between sp3-, sp2-, and sp-hybridized atomic orbitals on the main group elements (and 1s atomic orbitals on hydrogen), together with localized lone pairs derived from filled, nonbonding hybrid orbitals. The interaction that results in σ bonding takes the form of head-to-head overlap of the larger lobe of each hybrid orbital (or the single spherical lobe of a hydrogen 1s orbital). Each atomic orbital contributes one electron when the orbitals overlap pairwise to form two-electron σ bonds, or two electrons when the orbital constitutes a lone pair. These localized orbitals (bonding and non-bonding) are all located in the plane of the molecule, with σ bonds mainly localized between nuclei along the internuclear axis.
Pi (π) system or systems: Orthogonal to the σ framework described above, π bonding occurs above and below the plane of the molecule where σ bonding takes place. The π system(s) of the molecule are formed by the interaction of unhybridized p atomic orbitals on atoms employing sp2- and sp-hybridization. The interaction that results in π bonding takes place between p orbitals that are adjacent by virtue of a σ bond joining the atoms and takes the form of side-to-side overlap of the two equally large lobes that make up each p orbital. Atoms that are sp3-hybridized do not have an unhybridized p orbital available for participation in π bonding and their presence necessarily terminates a π system or separates two π systems. A basis p orbital that takes part in a π system can contribute one electron (which corresponds to half of a formal "double bond"), two electrons (which corresponds to a delocalized "lone pair"), or zero electrons (which corresponds to a formally "empty" orbital). Bonding for π systems formed from the overlap of more than two p orbitals is handled using the Hückel approach to obtain a zeroth order (qualitative) approximation of the π symmetry molecular orbitals that result from delocalized π bonding.
Using the σ/π-separation scheme to describe bonding, the Lewis resonance structures of a molecule like diazomethane can be translated into a bonding picture consisting of π-systems and localized lone pairs superimposed on a localized framework of σ-bonds.
This simple model for chemical bonding is successful for the description of most normal-valence molecules consisting of only s- and p-block elements, although systems that involve electron-deficient bonding, including nonclassical carbocations, lithium and boron clusters, and hypervalent centers require significant modifications in which σ bonds are also allowed to delocalize and are perhaps better treated with canonical molecular orbitals that are delocalized over the entire molecule. Likewise, d- and f-block organometallics are also inadequately described by this simple model. Bonds in strained small rings (such as cyclopropane or epoxide) are not well-described by strict σ/π separation, as bonding between atoms in the ring consists of "bent bonds" or "banana bonds" that are bowed outward and are intermediate in nature between σ and π bonds. Nevertheless, organic chemists frequently use the language of this model to rationalize the structure and reactivity of typical organic compounds.
Electrons in conjugated π systems are shared by all adjacent sp2- and sp-hybridized atoms that contribute overlapping, parallel p atomic orbitals. As such, the atoms and π-electrons involved behave as one large bonded system. These systems are often referred to 'n-center k-electron π-bonds,' compactly denoted by the symbol Πk n, to emphasize this behavior. For example, the delocalized π electrons in acetate anion and benzene are said to be involved in Π4 3 and Π6 6 systems, respectively (see the article on three-center four-electron bonding). Generally speaking, these multi-center bonds correspond to the occupation of several molecular orbitals (MOs) with varying degrees of bonding or non-bonding character (filling of orbitals with antibonding character is uncommon). Each one is occupied by one or two electrons in accordance with the Aufbau principle and Hund's rule. Cartoons showing overlapping p orbitals, like the one for benzene below, show the basis p atomic orbitals before they are combined to form molecular orbitals. In compliance with the Pauli exclusion principle, overlapping p orbitals do not result in the formation of one large MO containing more than two electrons.
Hückel MO theory is commonly used approach to obtain a zeroth order picture of delocalized π molecular orbitals, including the mathematical sign of the wavefunction at various parts of the molecule and the locations of nodal planes. It is particularly easy to apply for conjugated hydrocarbons and provides a reasonable approximation as long as the molecule is assumed to be planar with good overlap of p orbitals.
Stabilization energy
The quantitative estimation of stabilization from conjugation is notoriously contentious and depends on the implicit assumptions that are made when comparing reference systems or reactions. The energy of stabilization is known as the resonance energy when formally defined as the difference in energy between the real chemical species and the hypothetical species featuring localized π bonding that corresponds to the most stable resonance form.[6] This energy cannot be measured, and a precise definition accepted by most chemists will probably remain elusive. Nevertheless, some broad statements can be made. In general, stabilization is more significant for cationic systems than neutral ones. For buta-1,3-diene, a crude measure of stabilization is the activation energy for rotation of the C2-C3 bond. This places the resonance stabilization at around 6 kcal/mol.[7] Comparison of heats of hydrogenation of 1,4-pentadiene and 1,3-pentadiene estimates a slightly more modest value of 3.5 kcal/mol.[8] For comparison, allyl cation has a gas-phase rotation barrier of around 38 kcal/mol,[9] a much greater penalty for loss of conjugation. Comparison of hydride ion affinities of propyl cation and allyl cation, corrected for inductive effects, results in a considerably lower estimate of the resonance energy at 20–22 kcal/mol.[10] Nevertheless, it is clear that conjugation stabilizes allyl cation to a much greater extent than buta-1,3-diene. In contrast to the usually minor effect of neutral conjugation, aromatic stabilization can be considerable. Estimates for the resonance energy of benzene range from around 36–73 kcal/mol.[11]
Generalizations and related concepts
Homoconjugation weakens the double bond character of the C=O bond, resulting in a lower IR frequency.
There are also other types of interactions that generalize the idea of interacting p orbitals in a conjugated system. The concept of hyperconjugation holds that certain σ bonds can also delocalize into a low-lying unoccupied orbital of a π system or an unoccupied p orbital. Hyperconjugation is commonly invoked to explain the stability of alkyl substituted radicals and carbocations. Hyperconjugation is less important for species in which all atoms satisfy the octet rule, but a recent computational study supports hyperconjugation as the origin of the increased stability of alkenes with a higher degree of substitution (Zaitsev's rule).[12]
Homoconjugation[13] is an overlap of two π-systems separated by a non-conjugating group, such as CH2. Unambiguous examples are comparatively rare in neutral systems, due to a comparatively minor energetic benefit that is easily overridden by a variety of other factors; however, they are common in cationic systems in which a large energetic benefit can be derived from delocalization of positive charge (see the article on homoaromaticity for details.).[14] Neutral systems generally require constrained geometries favoring interaction to produce significant degrees of homoconjugation.[15] In the example below, the carbonyl stretching frequencies of the IR spectra of the respective compounds demonstrate homoconjugation, or lack thereof, in the neutral ground state molecules.
Due to the partial π character of formally σ bonds in a cyclopropane ring, evidence for transmission of "conjugation" through cyclopropanes has also been obtained.[16]
Two appropriately aligned π systems whose ends meet at right angles can engage in spiroconjugation[17] or in homoconjugation across the spiro atom.
Delocalization of negative charge in a generic carboxylate anion, derived from an organic carboxylic acid (cf. acetic acid), and the corresponding vinylogous carboxylate anion (the "vinylog/vinylogue" of the carboxylate anion), where a vinyl group now separates the charged oxygen from the carbonyl (C=O) group. The validity of the theoretical concept of vinylogy is supported by the pKa of such vinylogs, which approach that of the analogous carboxylic acid.
Vinylogy is the extension of a functional group through a conjugated organic bonding system, which transmits electronic effects.[18]
Conjugated cyclic compounds
Basis p orbitals of benzene.Benzene π molecular orbitals according to Hückel theory. Molecular orbitals are frequently described as linear combinations of atomic orbitals, whose coefficients are indicated here by the size and shading of the orbital lobes.
Cyclic compounds can be partly or completely conjugated. Annulenes, completely conjugated monocyclic hydrocarbons, may be aromatic, nonaromatic or antiaromatic.
Aromatic compounds
Compounds that have a monocyclic, planar conjugated system containing (4n + 2) π-electrons for whole numbers n are aromatic and exhibit an unusual stability. The classic example benzene has a system of six π electrons, which, together with the planar ring of C–C σ bonds containing 12electrons and radial C–H σ bonds containing six electrons, forms the thermodynamically and kinetically stable benzene ring, the common core of the benzenoid aromatic compounds. For benzene itself, there are two equivalent conjugated contributing Lewis structures (the so-called Kekulé structures) that predominate.[19][20] The true electronic structure is therefore a quantum-mechanical combination (resonance hybrid) of these contributors, which results in the experimentally observed C–C bonds which are intermediate between single and double bonds and of equal strength and length. In the molecular orbital picture, the six p atomic orbitals of benzene combine to give six molecular orbitals. Three of these orbitals, which lie at lower energies than the isolated p orbital and are therefore net bonding in character (one molecular orbital is strongly bonding, while the other two are equal in energy but bonding to a lesser extent) are occupied by six electrons, while three destabilized orbitals of overall antibonding character remain unoccupied. The result is strong thermodynamic and kinetic aromatic stabilization. Both models describe rings of π electron density above and below the framework of C–C σ bonds.
Nonaromatic and antiaromatic compounds
Cyclooctatetraene. Adjacent double bonds are not coplanar. The double bonds are therefore not conjugated.
Not all compounds with alternating double and single bonds are aromatic. Cyclooctatetraene, for example, possesses alternating single and double bonds. The molecule typically adopts a "tub" conformation. Because the p orbitals of the molecule do not align themselves well in this non-planar molecule, the π bonds are essentially isolated and not conjugated. The lack of conjugation allows the 8 π electron molecule to avoid antiaromaticity, a destabilizing effect associated with cyclic, conjugated systems containing 4n π (n = 0, 1, 2, ...) electrons. This effect is due to the placement of two electrons into two degenerate nonbonding (or nearly nonbonding) orbitals of the molecule, which, in addition to drastically reducing the thermodynamic stabilization of delocalization, would either force the molecule to take on triplet diradical character, or cause it to undergo Jahn-Teller distortion to relieve the degeneracy. This has the effect of greatly increasing the kinetic reactivity of the molecule. Because of the lack of long-range interactions, cyclooctatetraene takes on a nonplanar conformation and is nonaromatic in character, behaving as a typical alkene. In contrast, derivatives of the cyclooctatetraene dication and dianion have been found to be planar experimentally, in accord with the prediction that they are stabilized aromatic systems with 6 and 10 π electrons, respectively. Because antiaromaticity is a property that molecules try to avoid whenever possible, only a few experimentally observed species are believed to be antiaromatic. Cyclobutadiene and cyclopentadienyl cation are commonly cited as examples of antiaromatic systems.
In pigments
In a conjugated pi-system, electrons are able to capture certain photons as the electrons resonate along a certain distance of p-orbitals, similar to how a radio antenna detects radio signals along its length. Typically, the more conjugated (longer) the pi-system is, the longer the wavelength of photon can be captured. Compounds whose molecules contain a sufficient number of conjugated bonds can absorb light in the visible region, and therefore appear colorful to the eye, usually appearing yellow or red.[21]
Many dyes make use of conjugated electron systems to absorb visible light, giving rise to strong colors. For example, the long conjugated hydrocarbon chain in beta-carotene leads to its strong orange color. When an electron in the system absorbs a photon of light of the right wavelength, it can be promoted to a higher energy level. A simple model of the energy levels is provided by the quantum-mechanical problem of a one-dimensional particle in a box of length L, representing the movement of a π electron along a long conjugated chain of carbon atoms. In this model the lowest possible absorption energy corresponds to the energy difference between the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO). For a chain of n C=C bonds or 2n carbon atoms in the molecular ground state, there are 2n π electrons occupying n molecular orbitals, so that the energy gap is[22]
Since the box length L increases approximately linearly with the number of C=C bonds n, this means that the energy ΔE of a photon absorbed in the HOMO–LUMO transition is approximately proportional to 1/n. The photon wavelength λ = hc/ΔE is then approximately proportional to n. Although this model is very approximate, λ does in general increase with n (or L) for similar molecules. For example, the HOMO–LUMO absorption wavelengths for conjugated butadiene, hexatriene and octatetraene are 217nm, 252nm and 304nm respectively.[23] However, for good numerical agreement of the particle in a box model with experiment, the single-bond/double-bond bond length alternations of the polyenes must be taken into account.[24] Alternatively, one can use the Hückel method which is also designed to model the electronic structure of conjugated systems.
Many electronic transitions in conjugated π-systems are from a predominantly bondingmolecular orbital (MO) to a predominantly antibonding MO (π to π*), but electrons from non-bonding lone pairs can also be promoted to a π-system MO (n to π*) as often happens in charge-transfer complexes. A HOMO to LUMO transition is made by an electron if it is allowed by the selection rules for electromagnetic transitions. Conjugated systems of fewer than eight conjugated double bonds absorb only in the ultraviolet region and are colorless to the human eye. With every double bond added, the system absorbs photons of longer wavelength (and lower energy), and the compound ranges from yellow to red in color. Compounds that are blue or green typically do not rely on conjugated double bonds alone.
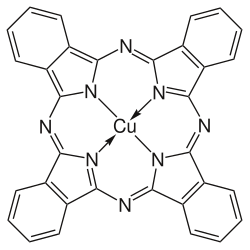
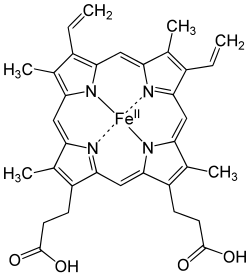
Porphyrins have conjugated molecular ring systems (macrocycles) that appear in many enzymes of biological systems. As a ligand, porphyrin forms numerous complexes with metallic ions like iron in hemoglobin that colors blood red. Hemoglobin transports oxygen to the cells of our bodies. Porphyrin–metal complexes often have strong colors. A similar molecular structural ring unit called chlorin is similarly complexed with magnesium instead of iron when forming part of the most common forms of chlorophyll molecules, giving them a green color. Another similar macrocycle unit is corrin, which complexes with cobalt when forming part of cobalamin molecules, constituting Vitamin B12, which is intensely red. The corrin unit has six conjugated double bonds but is not conjugated all the way around its macrocycle ring.
Conjugated systems form the basis of chromophores, which are light-absorbing parts of a molecule that can cause a compound to be colored. Such chromophores are often present in various organic compounds and sometimes present in polymers that are colored or glow in the dark. Chromophores often consist of a series of conjugated bonds and/or ring systems, commonly aromatic, which can include C–C, C=C, C=O, or N=N bonds.
Chemical structure of beta-carotene. The eleven conjugated double bonds that form the chromophore of the molecule are highlighted in red.
↑For the purposes of this article, we are primarily concerned with delocalized orbitals with π-symmetry. This is in line with the typical usage of 'conjugated system' to refer to π (and not σ) delocalization. Canonical molecular orbitals are fully delocalized, so in a sense, all electrons involved in bonding, including ones making up the σ bonds and lone pairs, are delocalized throughout the molecule. However, while treating π electrons as delocalized yields many useful insights into chemical reactivity, treatment of σ and nonbonding electrons in the same way is generally less profitable, except in cases of multicenter σ-bonding as found in cluster compounds of Li and B. Moreover, the added complexity tends to impede chemical intuition. Hence, for most organic molecules, chemists commonly use a localized orbital model to describe the σ-bonds and lone pairs, while superimposing delocalized molecular orbitals to describe the π-bonding. This view has the added advantage that there is a clear correspondence between the Lewis structure of a molecule and the orbitals used to describe its bonding.
References
↑Thiele, Johannes (1899). "Zur Kenntnis der ungesättigten Verbindungen"[[Contribution] to our knowledge of unsaturated compounds]. Justus Liebig's Annalen der Chemie (in German). 306: 87–142. doi:10.1002/jlac.18993060107. On p. 90, Thiele coined the term "conjugated": "Ein solches System benachbarter Doppelbindungen mit ausgeglichenen inneren Partialvalenzen sei als 'conjugirt' bezeichnet." (Such a system of adjacent double bonds with equalized inner partial valences shall be termed "conjugated".)
↑Feller, David; Craig, Norman C. (2009-02-05). "High Level ab Initio Energies and Structures for the Rotamers of 1,3-Butadiene". The Journal of Physical Chemistry A. 113 (8): 1601–1607. Bibcode:2009JPCA..113.1601F. doi:10.1021/jp8095709. PMID19199679.
↑Carey, Francis A.; Guiliano, Robert M. (2013-01-07). Organic chemistry (Ninthed.). New York, NY. ISBN9780073402741. OCLC822971422.{{cite book}}: CS1 maint: location missing publisher (link)
↑Barbour, Josiah B.; Karty, Joel M. (2004-01-14). "Resonance Energies of the Allyl Cation and Allyl Anion: Contribution by Resonance and Inductive Effects toward the Acidity and Hydride Abstraction Enthalpy of Propene". The Journal of Organic Chemistry. 69 (3): 648–654. doi:10.1021/jo035189m. PMID14750787.
↑Braida, Benoit; Prana, Vinca; Hiberty, Philippe C. (2009-07-20). "The Physical Origin of Saytzeff's Rule". Angewandte Chemie International Edition. 48 (31): 5724–5728. doi:10.1002/anie.200901923. ISSN1433-7851. PMID19562814.
↑Some orbital overlap is possible even between bonds separated by one (or more) CH2 because the bonding electrons occupy orbitals which are quantum-mechanical functions and extend indefinitely in space. Macroscopic drawings and models with sharp boundaries are misleading because they do not show this aspect.
↑Stewart, John Mathews; Pagenkopf, Gordon K. (January 1969). "Transmission of conjugation by the cyclopropane ring". The Journal of Organic Chemistry. 34 (1): 7–11. doi:10.1021/jo00838a003. ISSN0022-3263.
↑The Vinylogous Aldol Reaction: A Valuable, Yet Understated Carbon-Carbon Bond-Forming Maneuver Giovanni Casiraghi, Franca Zanardi, Giovanni Appendino, and Gloria Rassu Chem. Rev.2000; 100(6) pp 1929 – 1972; (Review) doi:10.1021/cr990247i
↑Rashid, Zahid; van Lenthe, Joop H. (March 2011). "Generation of Kekulé valence structures and the corresponding valence bond wave function". Journal of Computational Chemistry. 32 (4): 696–708. doi:10.1002/jcc.21655. ISSN1096-987X. PMID20941739. S2CID16526798.
↑While the two Kekulé resonance forms contribute to most (>90%) of the π bond energy, there are also a number of other minor contributors to the wavefunction in the valence bond treatment, including the three Dewar resonance forms, and even smaller contributions from various ionic and singlet diradical forms. See article by Rashid and van Lenthe for a recent computational treatment.
↑Autschbach, Jochen (November 2007). "Why the Particle-in-a-Box Model Works Well for Cyanine Dyes but Not for Conjugated Polyenes". Journal of Chemical Education. 84 (11): 1840. Bibcode:2007JChEd..84.1840A. doi:10.1021/ed084p1840. ISSN0021-9584.
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.