The term dipolar bond is used in organic chemistry for compounds such as amine oxides for which the electronic structure can be described in terms of the basic amine donating two electrons to an oxygen atom.
R 3N → O
The arrow → indicates that both electrons in the bond originate from the amine moiety. In a standard covalent bond each atom contributes one electron. Therefore, an alternative description is that the amine gives away one electron to the oxygen atom, which is then used, with the remaining unpaired electron on the nitrogen atom, to form a standard covalent bond. The process of transferring the electron from nitrogen to oxygen creates formal charges, so the electronic structure may also be depicted as
R 3N+ O−
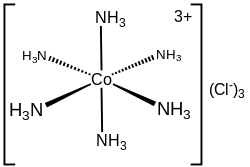
Hexamminecobalt(III) chloride
This electronic structure has an electric dipole, hence the name polar bond. In reality, the atoms carry partial charges; the more electronegative atom of the two involved in the bond will usually carry a partial negative charge. One exception to this is carbon monoxide. In this case, the carbon atom carries the partial negative charge although it is less electronegative than oxygen.
An example of a dative covalent bond is provided by the interaction between a molecule of ammonia, a Lewis base with a lone pair of electrons on the nitrogen atom, and boron trifluoride, a Lewis acid by virtue of the boron atom having an incomplete octet of electrons. In forming the adduct, the boron atom attains an octet configuration.
The electronic structure of a coordination complex can be described in terms of the set of ligands each donating a pair of electrons to a metal centre. For example, in hexamminecobalt(III) chloride, each ammonia ligand donates its lone pair of electrons to the cobalt(III) ion. In this case, the bonds formed are described as coordinate bonds. In the Covalent Bond Classification (CBC) method, ligands that form coordinate covalent bonds with a central atom are classed as L-type, while those that form normal covalent bonds are classed as X-type.
Comparison with other electron-sharing modes
In all cases, the bond, whether dative or "normal" electron-sharing, is a covalent bond. In common usage, the prefix dipolar, dative or coordinate merely serves to indicate the origin of the electrons used in creating the bond. For example, F3B ← O(C2H5)2 ("boron trifluoride (diethyl) etherate") is prepared from BF3 and :O(C2H5)2, as opposed to the radical species [•BF3]– and [•O(C2H5)2]+. The dative bond is also a convenience in terms of notation, as formal charges are avoided: we can write D: + []A ⇌ D → A rather than D+–A– (here : and [] represent the lone-pair and empty orbital on the electron-pair donor D and acceptor A, respectively). The notation is sometimes used even when the Lewis acid-base reaction involved is only notional (e.g., the sulfoxide R2S → O is rarely if ever made by reacting the sulfide R2S with atomic oxygen O). Thus, most chemists do not make any claim with respect to the properties of the bond when choosing one notation over the other (formal charges vs. arrow bond).
It is generally true, however, that bonds depicted this way are polar covalent, sometimes strongly so, and some authors claim that there are genuine differences in the properties of a dative bond and electron-sharing bond and suggest that showing a dative bond is more appropriate in particular situations. As far back as 1989, Haaland characterized dative bonds as bonds that are (i) weak and long; (ii) with only a small degree of charge-transfer taking place during bond formation; and (iii) whose preferred mode of dissociation in the gas phase (or low ε inert solvent) is heterolytic rather than homolytic.[7] The ammonia-borane adduct (H3N → BH3) is given as a classic example: the bond is weak, with a dissociation energy of 31 kcal/mol (cf. 90 kcal/mol for ethane), and long, at 166 pm (cf. 153 pm for ethane), and the molecule possesses a dipole moment of 5.2 D that implies a transfer of only 0.2 e– from nitrogen to boron. The heterolytic dissociation of H3N → BH3 is estimated to require 27 kcal/mol, confirming that heterolysis into ammonia and borane is more favorable than homolysis into radical cation and radical anion. However, aside from clear-cut examples, there is considerable dispute as to when a particular compound qualifies and, thus, the overall prevalence of dative bonding (with respect to an author's preferred definition). Computational chemists have suggested quantitative criteria to distinguish between the two "types" of bonding.[8][9][10]
Some non-obvious examples where dative bonding is claimed to be important include carbon suboxide (O≡C → C0 ← C≡O), tetraaminoallenes (described using dative bond language as "carbodicarbenes"; (R2N)2C → C0 ← C(NR2)2), the Ramirez carbodiphosphorane (Ph3P → C0 ← PPh3), and bis(triphenylphosphine)iminium cation (Ph3P → N+ ← PPh3), all of which exhibit considerably bent equilibrium geometries, though with a shallow barrier to bending. Simple application of the normal rules for drawing Lewis structures by maximizing bonding (using electron-sharing bonds) and minimizing formal charges would predict heterocumulene structures, and therefore linear geometries, for each of these compounds. Thus, these molecules are claimed to be better modeled as coordination complexes of :C: (carbon(0) or carbone) or :N:+ (mononitrogen cation) with CO, PPh3, or N-heterocycliccarbenes as ligands, the lone-pairs on the central atom accounting for the bent geometry. However, the usefulness of this view is disputed.[9][10]
↑Haaland, Arne (1989). "Covalent versus Dative Bonds to Main Group Metals, a Useful Distinction". Angewandte Chemie International Edition in English. 28 (8): 992–1007. doi:10.1002/anie.198909921. ISSN0570-0833.
↑Himmel, Daniel; Krossing, Ingo; Schnepf, Andreas (2014-01-07). "Dative Bonds in Main-Group Compounds: A Case for Fewer Arrows!". Angewandte Chemie International Edition. 53 (2): 370–374. Bibcode:2014ACIE...53..370H. doi:10.1002/anie.201300461. PMID24243854.
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.