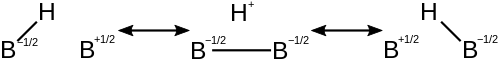In chemistry, resonance, also called mesomerism, is a way of describing bonding in certain molecules or polyatomic ions by the combination of several contributing structures (or forms,[1] also variously known as resonance structures or canonical structures) into a resonance hybrid (or hybrid structure) in valence bond theory. It has particular value for analyzing delocalized electrons where the bonding cannot be expressed by one single Lewis structure. The resonance hybrid is the accurate structure for a molecule or ion; it is an average of the theoretical (or hypothetical) contributing structures.
Under the framework of valence bond theory, resonance is an extension of the idea that the bonding in a chemical species can be described by a Lewis structure. For many chemical species, a single Lewis structure, consisting of atoms obeying the octet rule, possibly bearing formal charges, and connected by bonds of positive integer order, is sufficient for describing the chemical bonding and rationalizing experimentally determined molecular properties like bond lengths, angles, and dipole moment.[2] However, in some cases, more than one Lewis structure could be drawn, and experimental properties are inconsistent with any one structure. In order to address this type of situation, several contributing structures are considered together as an average, and the molecule is said to be represented by a resonance hybrid in which several Lewis structures are used collectively to describe its true structure.
The experimental geometry of the nitrite anion, NO2 , shown on the right, is best rationalized by describing its structure as a resonance hybrid consisting of two major and equally important contributing forms.
For instance, in NO2–, nitrite anion, the two N–O bond lengths are equal, even though no single Lewis structure has two N–O bonds with the same formal bond order. However, its measured structure is consistent with a description as a resonance hybrid of the two major contributing structures shown above: it has two equal N–O bonds of 125 pm, intermediate in length between a typical N–O single bond (145 pm in hydroxylamine, H2N–OH) and N–O double bond (115 pm in nitronium ion, [O=N=O]+). According to the contributing structures, each N–O bond is an average of a formal single and formal double bond, leading to a true bond order of 1.5. By virtue of this averaging, the Lewis description of the bonding in NO2– is reconciled with the experimental fact that the anion has equivalent N–O bonds.
The resonance hybrid represents the actual molecule as the "average" of the contributing structures, with bond lengths and partial charges taking on intermediate values compared to those expected for the individual Lewis structures of the contributors, were they to exist as "real" chemical entities.[3] The contributing structures differ only in the formal apportionment of electrons to the atoms, and not in the actual physically and chemically significant electron or spin density. While contributing structures may differ in formal bond orders and in formal charge assignments, all contributing structures must have the same number of valence electrons and the same spin multiplicity.[4]
Because electron delocalization lowers the potential energy of a system, any species represented by a resonance hybrid is more stable than any of the (hypothetical) contributing structures.[5] Electron delocalization stabilizes a molecule because the electrons are more evenly spread out over the molecule, decreasing electron-electron repulsion.[6] The difference in potential energy between the actual species and the (computed) energy of the contributing structure with the lowest potential energy is called the resonance energy[7] or delocalization energy. The magnitude of the resonance energy depends on assumptions made about the hypothetical "non-stabilized" species and the computational methods used and does not represent a measurable physical quantity, although comparisons of resonance energies computed under similar assumptions and conditions may be chemically meaningful.
Molecules with an extended π system such as linear polyenes and polyaromatic compounds are well described by resonance hybrids as well as by delocalized orbitals in molecular orbital theory.
Resonance vs isomerism
Resonance is to be distinguished from isomerism. Isomers are molecules with the same chemical formula but are distinct chemical species with different arrangements of atomic nuclei in space. Resonance contributors of a molecule, on the other hand, can only differ in the way electrons are formally assigned to atoms in the Lewis structure depictions of the molecule. Specifically, when a molecular structure is said to be represented by a resonance hybrid, it does not mean that electrons of the molecule are "resonating" or shifting back and forth between several sets of positions, each one represented by a Lewis structure. Rather, it means that the set of contributing structures represents an intermediate structure (a weighted average of the contributors), with a single, well-defined geometry and distribution of electrons. It is incorrect to regard resonance hybrids as rapidly interconverting isomers, even though the term "resonance" might evoke such an image.[8] (As described below, the term "resonance" originated as a classical physics analogy for a quantum mechanical phenomenon, so it should not be construed too literally.) Symbolically, the double headed arrow is used to indicate that A and B are contributing forms of a single chemical species (as opposed to an equilibrium arrow, e.g., ; see below for details on usage).
A non-chemical analogy is illustrative: one can describe the characteristics of a real animal, the narwhal, in terms of the characteristics of two mythical creatures: the unicorn, a creature with a single horn on its head, and the leviathan, a large, whale-like creature. The narwhal is not a creature that goes back and forth between being a unicorn and being a leviathan, nor do the unicorn and leviathan have any physical existence outside the collective human imagination. Nevertheless, describing the narwhal in terms of these imaginary creatures provides a reasonably good description of its physical characteristics.
Due to confusion with the physical meaning of the word resonance, as no entities actually physically "resonate", it has been suggested that the term resonance be abandoned in favor of delocalization[9] and resonance energy abandoned in favor of delocalization energy. A resonance structure becomes a contributing structure and the resonance hybrid becomes the hybrid structure. The double headed arrows would be replaced by commas to illustrate a set of structures, as arrows of any type may suggest that a chemical change is taking place.
Representation in diagrams
Contributing structures of the thiocyanate ion, enclosed in square brackets.
In diagrams, contributing structures are typically separated by double-headed arrows (↔). The arrow should not be confused with the right and left pointing equilibrium arrow (⇌). All structures together may be enclosed in large square brackets, to indicate they picture one single molecule or ion, not different species in a chemical equilibrium.
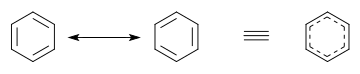
Alternatively to the use of contributing structures in diagrams, a hybrid structure can be used. In a hybrid structure, pi bonds that are involved in resonance are usually pictured as curves[10] or dashed lines, indicating that these are partial rather than normal complete pi bonds. In benzene and other aromatic rings, the delocalized pi-electrons are sometimes pictured as a solid circle.[11]
History
The concept first appeared in 1899 in Johannes Thiele's "Partial Valence Hypothesis" to explain the unusual stability of benzene which would not be expected from August Kekulé's structure proposed in 1865 with alternating single and double bonds.[12] Benzene undergoes substitution reactions, rather than addition reactions as typical for alkenes. He proposed that the carbon-carbon bond in benzene is intermediate of a single and double bond.
The resonance proposal also helped explain the number of isomers of benzene derivatives. For example, Kekulé's structure would predict four dibromobenzene isomers, including two ortho isomers with the brominated carbon atoms joined by either a single or a double bond. In reality there are only three dibromobenzene isomers and only one is ortho, in agreement with the idea that there is only one type of carbon-carbon bond, intermediate between a single and a double bond.[13]
The mechanism of resonance was introduced into quantum mechanics by Werner Heisenberg in 1926 in a discussion of the quantum states of the helium atom. He compared the structure of the helium atom with the classical system of resonating coupled harmonic oscillators.[3][14] In the classical system, the coupling produces two modes, one of which is lower in frequency than either of the uncoupled vibrations; quantum mechanically, this lower frequency is interpreted as a lower energy. Linus Pauling used this mechanism to explain the partial valence of molecules in 1928, and developed it further in a series of papers in 1931-1933.[15][16] The alternative term mesomerism[17] popular in German and French publications with the same meaning was introduced by C. K. Ingold in 1938, but did not catch on in the English literature. The current concept of mesomeric effect has taken on a related but different meaning. The double headed arrow was introduced by the German chemist Fritz Arndt who preferred the German phrase zwischenstufe or intermediate stage.
Resonance theory dominated over competing Hückel method for two decades thanks to being relatively easier to understand for chemists without fundamental physics background, even if they couldn't grasp the concept of quantum superposition and confused it with tautomerism. Pauling and Wheland themselves characterized Erich Hückel's approach as "cumbersome" at the time, and his lack of communication skills contributed: when Robert Robinson sent him a friendly request, he responded arrogantly that he is not interested in organic chemistry.[18]
In the Soviet Union, resonance theory – especially as developed by Pauling – was attacked in the early 1950s as being contrary to the Marxist principles of dialectical materialism, and in June 1951 the Soviet Academy of Sciences under the leadership of Alexander Nesmeyanov convened a conference on the chemical structure of organic compounds, attended by 400 physicists, chemists, and philosophers, where "the pseudo-scientific essence of the theory of resonance was exposed and unmasked".[19]
Major and minor contributors
One contributing structure may resemble the actual molecule more than another (in the sense of energy and stability). Structures with a low value of potential energy are more stable than those with high values and resemble the actual structure more. The most stable contributing structures are called major contributors. Energetically unfavourable and therefore less favorable structures are minor contributors. With rules listed in rough order of diminishing importance, major contributors are generally structures that
obey as much as possible the octet rule (8 valence electrons around each atom rather than having deficiencies or surplus, or 2 electrons for Period 1 elements);
place negative charge, if any, on the most electronegative atoms and positive charge, if any, on the most electropositive;
do not deviate substantially from idealized bond lengths and angles (e.g., the relative unimportance of Dewar-type resonance contributors for benzene);
maintain aromatic substructures locally while avoiding anti-aromatic ones (seeClar sextetandbiphenylene).
A maximum of eight valence electrons is strict for the Period 2 elements Be, B, C, N, O, and F, as is a maximum of two for H and He and effectively for Li as well.[20] The issue of expansion of the valence shell of third period and heavier main group elements is controversial. A Lewis structure in which a central atom has a valence electron count greater than eight traditionally implies the participation of d orbitals in bonding. However, the consensus opinion is that while they may make a marginal contribution, the participation of d orbitals is unimportant, and the bonding of so-called hypervalent molecules are, for the most part, better explained by charge-separated contributing forms that depict three-center four-electron bonding. Nevertheless, by tradition, expanded octet structures are still commonly drawn for functional groups like sulfoxides, sulfones, and phosphorus ylides, for example. Regarded as a formalism that does not necessarily reflect the true electronic structure, such depictions are preferred by the IUPAC over structures featuring partial bonds, charge separation, or dative bonds.[21]
Equivalent contributors contribute equally to the actual structure, while the importance of nonequivalent contributors is determined by the extent to which they conform to the properties listed above. A larger number of significant contributing structures and a more voluminous space available for delocalized electrons lead to stabilization (lowering of the energy) of the molecule.
In benzene the two cyclohexatriene Kekulé structures, first proposed by Kekulé, are taken together as contributing structures to represent the total structure. In the hybrid structure on the right, the dashed hexagon replaces three double bonds, and represents six electrons in a set of three molecular orbitals of π symmetry, with a nodal plane in the plane of the molecule.
In furan a lone pair of the oxygen atom interacts with the π orbitals of the carbon atoms. The curved arrows depict the permutation of delocalized π electrons, which results in different contributors.
Electron-rich molecules
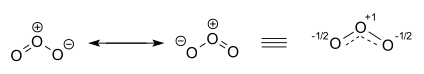
The ozone molecule is represented by two contributing structures. In reality the two terminal oxygen atoms are equivalent and the hybrid structure is drawn on the right with a charge of −1⁄2 on both oxygen atoms and partial double bonds with a full and dashed line and bond order1+1⁄2.[22][23]
For hypervalent molecules, the rationalization described above can be applied to generate contributing structures to explain the bonding in such molecules. Shown below are the contributing structures of a 3c-4e bond in xenon difluoride.
The allyl cation has two contributing structures with a positive charge on the terminal carbon atoms. In the hybrid structure their charge is +1⁄2. The full positive charge can also be depicted as delocalized among three carbon atoms.
The diborane molecule is described by contributing structures, each with electron-deficiency on different atoms. This reduces the electron-deficiency on each atom and stabilizes the molecule. Below are the contributing structures of an individual 3c-2e bond in diborane.
Often, reactive intermediates such as carbocations and free radicals have more delocalized structure than their parent reactants, giving rise to unexpected products. The classical example is allylic rearrangement.[24] When 1 mole of HCl adds to 1 mole of 1,3-butadiene, in addition to the ordinarily expected product 3-chloro-1-butene, we also find 1-chloro-2-butene. Isotope labelling experiments have shown that what happens here is that the additional double bond shifts from 1,2 position to 2,3 position in some of the product. This and other evidence (such as NMR in superacid solutions) shows that the intermediate carbocation must have a highly delocalized structure, different from its mostly classical (delocalization exists but is small) parent molecule. This cation (an allylic cation) can be represented using resonance, as shown above.
This observation of greater delocalization in less stable molecules is quite general. The excited states of conjugated dienes are stabilized more by conjugation than their ground states, causing them to become organic dyes.[25]
A well-studied example of delocalization that does not involve π electrons (hyperconjugation) can be observed in the non-classical 2-Norbornyl cation[26] Another example is methanium (CH+ 5). These can be viewed as containing three-center two-electron bonds and are represented either by contributing structures involving rearrangement of σ electrons or by a special notation, a Y that has the three nuclei at its three points.
Delocalized electrons are important for several reasons; a major one is that an expected chemical reaction may not occur because the electrons delocalize to a more stable configuration, resulting in a reaction that happens at a different location. An example is the Friedel–Craftsalkylation[27] of benzene with 1-chloro-2-methylpropane; the carbocation rearranges to a tert-butyl group stabilized by hyperconjugation, a particular form of delocalization.
Comparing the two contributing structures of benzene, all single and double bonds are interchanged. Bond lengths can be measured, for example using X-ray diffraction. The average length of a C–C single bond is 154 pm; that of a C=C double bond is 133pm. In localized cyclohexatriene, the carbon–carbon bonds should be alternating 154 and 133pm. Instead, all carbon–carbon bonds in benzene are found to be about 139pm, a bond length intermediate between single and double bond. This mixed single and double bond (or triple bond) character is typical for all molecules in which bonds have a different bond order in different contributing structures. Bond lengths can be compared using bond orders. For example, in cyclohexane the bond order is 1 while that in benzene is 1+(3÷6) = 1+1⁄2. Consequently, benzene has more double bond character and hence has a shorter bond length than cyclohexane.
Resonance energy
Resonance (or delocalization) energy is the amount of energy needed to convert the true delocalized structure into that of the most stable contributing structure. The empirical resonance energy can be estimated by comparing the enthalpy change of hydrogenation of the real substance with that estimated for the contributing structure.
Hydrogenation of one mole of double bonds delivers 119.7kJ (28.6kcal), as can be deduced from the last step, the hydrogenation of cyclohexene. In benzene, however, 23.4kJ (5.6kcal) are needed to hydrogenate one mole of double bonds. The difference, being 143.1kJ (34.2kcal), is the empirical resonance energy of benzene. Because 1,3-cyclohexadiene also has a small delocalization energy (7.6kJ or 1.8kcal/mol) the net resonance energy, relative to the localized cyclohexatriene, is a bit higher: 151kJ or 36kcal/mol. [28]
This measured resonance energy is also the difference between the hydrogenation energy of three 'non-resonance' double bonds and the measured hydrogenation energy:
Regardless of their exact values, resonance energies of various related compounds provide insights into their bonding. The resonance energies for pyrrole, thiophene, and furan are, respectively, 88, 121, and 67 kJ/mol (21, 29, and 16 kcal/mol).[30] Thus, these heterocycles are far less aromatic than benzene, as is manifested in the lability of these rings.
Quantum mechanical description in valence bond (VB) theory
VB mixing diagram of benzene. The A1g and B2u labels define the symmetries of the two states, as defined by the character table for the D6hsymmetry group.
Resonance has a deeper significance in the mathematical formalism of valence bond theory (VB). Quantum mechanics requires that the wavefunction of a molecule obey its observed symmetry. If a single contributing structure does not achieve this, resonance is invoked.
For example, in benzene, valence bond theory begins with the two Kekulé structures which do not individually possess the sixfold symmetry of the real molecule. The theory constructs the actual wave function as a linear superposition of the wave functions representing the two structures. As both Kekulé structures have equal energy, they are equal contributors to the overall structure – the superposition is an equally weighted average, or a 1:1 linear combination of the two in the case of benzene. The symmetric combination gives the ground state, while the antisymmetric combination gives the first excited state, as shown.
In general, the superposition is written with undetermined coefficients, which are then variationallyoptimized to find the lowest possible energy for the given set of basis wave functions. When more contributing structures are included, the molecular wave function becomes more accurate and more excited states can be derived from different combinations of the contributing structures.
Comparison with molecular orbital (MO) theory
π molecular orbitals of benzene
In molecular orbital theory, the main alternative to valence bond theory, the molecular orbitals (MOs) are approximated as sums of all the atomic orbitals (AOs) on all the atoms; there are as many MOs as AOs. Each AOi has a weighting coefficient ci that indicates the AO's contribution to a particular MO. For example, in benzene, the MO model gives us 6 π MOs which are combinations of the 2pz AOs on each of the 6 C atoms. Thus, each π MO is delocalized over the whole benzene molecule and any electron occupying an MO will be delocalized over the whole molecule. This MO interpretation has inspired the picture of the benzene ring as a hexagon with a circle inside. When describing benzene, the VB concept of localized σ bonds and the MO concept of delocalized π orbitals are frequently combined in elementary chemistry courses.
The contributing structures in the VB model are particularly useful in predicting the effect of substituents on π systems such as benzene. They lead to the models of contributing structures for an electron-withdrawing group and electron-releasing group on benzene. The utility of MO theory is that a quantitative indication of the charge from the π system on an atom can be obtained from the squares of the weighting coefficient ci on atom Ci. Charge qi≈c2 i. The reason for squaring the coefficient is that if an electron is described by an AO, then the square of the AO gives the electron density. The AOs are adjusted (normalized) so that AO2=1, and qi≈(ciAOi)2 ≈c2 i. In benzene, qi=1 on each C atom. With an electron-withdrawing groupqi<1 on the ortho and para C atoms and qi>1 for an electron-releasing group.
Coefficients
Weighting of the contributing structures in terms of their contribution to the overall structure can be calculated in multiple ways, using "Ab initio" methods derived from Valence Bond theory, or else from the Natural Bond Orbitals (NBO) approaches of Weinhold NBO5Archived 2008-02-08 at the Wayback Machine , or finally from empirical calculations based on the Hückel method. A Hückel method-based software for teaching resonance is available on the HuLiS Web site.
Charge delocalization
In the case of ions it is common to speak about delocalized charge (charge delocalization). An example of delocalized charge in ions can be found in the carboxylate group, wherein the negative charge is centered equally on the two oxygen atoms. Charge delocalization in anions is an important factor determining their reactivity (generally: the higher the extent of delocalization the lower the reactivity) and, specifically, the acid strength of their conjugate acids. As a general rule, the better delocalized is the charge in an anion the stronger is its conjugate acid. For example, the negative charge in perchlorate anion (ClO− 4) is evenly distributed among the symmetrically oriented oxygen atoms (and a part of it is also kept by the central chlorine atom). This excellent charge delocalization combined with the high number of oxygen atoms (four) and high electronegativity of the central chlorine atom leads to perchloric acid being one of the strongest known acids with a pKa value of −10.[32] The extent of charge delocalization in an anion can be quantitatively expressed via the WAPS (weighted average positive sigma) parameter[33] parameter and an analogous WANS (weighted average negative sigma)[34][35] parameter is used for cations.
WAPS values of anions of common acids and WANS values of cations of common bases
1 2 Pauling, Linus (1960). "The Concept of Resonance". The Nature of the Chemical Bond – An Introduction to Modern Structural Chemistry (3rded.). Cornell University Press. pp.10–13. ISBN978-0801403330.{{cite book}}: ISBN / Date incompatibility (help)
↑ Practicing chemists familiar with the concepts of resonance and delocalization will often draw just one major contributing structure to implicitly represent a molecule whose structure should be described by invoking a resonance hybrid. For example, a chemist might arbitrarily choose to draw the resonance contributor of NO2– shown on the left, with the understanding that the reader is aware of the other contributor, shown on the right, as well as the implication that the N–O bonds are actually equivalent. This practice is especially prevalent in organic chemistry, where one of the Kekulé structures of benzene is frequently chosen to depict the regular hexagonal structure of the molecule.
↑ Morrison, Robert; Boyd, Robert (1989). "Chapter 10". Organic Chemistry (5thed.). Prentice Hall of India. p.372. ISBN978-0-87692-560-7. The resonance hybrid is more stable than any of the contributing structures.
↑ Carey, Francis A.; Sundberg, Richard J. (2007). Advanced Organic Chemistry Part A: Structure and Mechanisms. Springer. p.19. ISBN978-0-387-68346-1.
↑ Thiele, Johannes (1899). "Zur Kenntnis der ungesättigten Verbindungen"[[Contribution] to our knowledge of unsaturated compounds]. Justus Liebig's Annalen der Chemie (in German). 306: 87–142. doi:10.1002/jlac.18993060107. On p. 89, Thiele introduced the concept of "partial valence": "Ich nehme nun an, ... eine Partialvalens vorhanden ist, eine Annahme, die sich auch thermisch begründen lässt." (Now I assume that in the case of substances to which a double bond is attributed, actually two affinities of each of the participating atoms are used for their bond; however, on account of the capacity for addition of double bonds, the power of affinity is not completely consumed, and in each of the atoms a remnant of affinity or a "partial valence" exists – an assumption that can also be substantiated thermally [i.e., via calorimetry].) On p. 90, Thiele coined the term "conjugated": "Ein solches System benachbarter Doppelbindungen mit ausgeglichenen inneren Partialvalenzen sei als conjugirt bezeichnet." (Such a system of adjacent double bonds with equalized inner partial valences shall be termed "conjugated".) Thiele discussed the conjugated structure of benzene on pp. 125–129: VIII. Die aromatischen Verbindungen. Das Benzol. (VIII. The aromatic compounds. Benzene.)
↑ Pauling, L. (1960). The Nature of the Chemical Bond (3rded.). Oxford University Press. p.184. In this source, Pauling first mentions related papers by Slater and Hückel in 1931, and then cites his own key papers: Pauling, Linus. (1931). "The Nature of the Chemical Bond. Ii. The One-Electron Bond and the Three-Electron Bond". J. Am. Chem. Soc. 53 (1367): 3225. Bibcode:1931JAChS..53.3225P. doi:10.1021/ja01360a004. and subsequent papers in 1932–33.
↑ Moore, Barrington Jr. (1954). Terror and Progress USSR: Some Sources of Change and Stability in the Soviet Dictatorship. pp.142–143.
↑ Lithium is always found as Li+ (1s2), a duet, in ionic compounds. In compounds like CH3Li with some degree of covalency, bonding is achieved primarily with the 2s orbital, with some contribution from a 2p orbital. (This bonding scheme is used in condensed phase aggregates like (CH3Li)4 as well, leading to a higher coordination number for lithium.) Thus, in principle, up to an octet can be accommodated. Nevertheless, the formal number of valence electrons around Li never exceeds two, unless weak donor-acceptor interactions with neutral ligands (e.g., solvent molecules, often omitted from Lewis structures) are included.
↑ Wiberg; Nakaji; Morgan (1993). "Heat of hydrogenation of a cis imine. An experimental and theoretical study". J. Am. Chem. Soc. 115 (9): 3527–3532. Bibcode:1993JAChS.115.3527W. doi:10.1021/ja00062a017.
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.