
A covalent bond is a chemical bond that involves the sharing of electrons to form electron pairs between atoms. These electron pairs are known as shared pairs or bonding pairs. The stable balance of attractive and repulsive forces between atoms, when they share electrons, is known as covalent bonding. For many molecules, the sharing of electrons allows each atom to attain the equivalent of a full valence shell, corresponding to a stable electronic configuration. In organic chemistry, covalent bonding is much more common than ionic bonding.

Diatomic molecules are molecules composed of only two atoms, of the same or different chemical elements. If a diatomic molecule consists of two atoms of the same element, such as hydrogen or oxygen, then it is said to be homonuclear. Otherwise, if a diatomic molecule consists of two different atoms, such as carbon monoxide or nitric oxide, the molecule is said to be heteronuclear. The bond in a homonuclear diatomic molecule is non-polar.
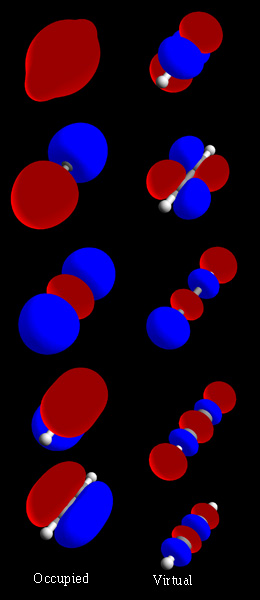
In chemistry, a molecular orbital is a mathematical function describing the location and wave-like behavior of an electron in a molecule. This function can be used to calculate chemical and physical properties such as the probability of finding an electron in any specific region. The terms atomic orbital and molecular orbital were introduced by Robert S. Mulliken in 1932 to mean one-electron orbital wave functions. At an elementary level, they are used to describe the region of space in which a function has a significant amplitude.

A quantum mechanical system or particle that is bound—that is, confined spatially—can only take on certain discrete values of energy, called energy levels. This contrasts with classical particles, which can have any amount of energy. The term is commonly used for the energy levels of the electrons in atoms, ions, or molecules, which are bound by the electric field of the nucleus, but can also refer to energy levels of nuclei or vibrational or rotational energy levels in molecules. The energy spectrum of a system with such discrete energy levels is said to be quantized.

In theoretical chemistry, a conjugated system is a system of connected p-orbitals with delocalized electrons in a molecule, which in general lowers the overall energy of the molecule and increases stability. It is conventionally represented as having alternating single and multiple bonds. Lone pairs, radicals or carbenium ions may be part of the system, which may be cyclic, acyclic, linear or mixed. The term "conjugated" was coined in 1899 by the German chemist Johannes Thiele.

In chemistry, pi bonds are covalent chemical bonds, in each of which two lobes of an orbital on one atom overlap with two lobes of an orbital on another atom, and in which this overlap occurs laterally. Each of these atomic orbitals has an electron density of zero at a shared nodal plane that passes through the two bonded nuclei. This plane also is a nodal plane for the molecular orbital of the pi bond. Pi bonds can form in double and triple bonds but do not form in single bonds in most cases.
In chemistry, molecular orbital theory is a method for describing the electronic structure of molecules using quantum mechanics. It was proposed early in the 20th century.
In chemistry, valence bond (VB) theory is one of the two basic theories, along with molecular orbital (MO) theory, that were developed to use the methods of quantum mechanics to explain chemical bonding. It focuses on how the atomic orbitals of the dissociated atoms combine to give individual chemical bonds when a molecule is formed. In contrast, molecular orbital theory has orbitals that cover the whole molecule.

In chemistry, sigma bonds are the strongest type of covalent chemical bond. They are formed by head-on overlapping between atomic orbitals. Sigma bonding is most simply defined for diatomic molecules using the language and tools of symmetry groups. In this formal approach, a σ-bond is symmetrical with respect to rotation about the bond axis. By this definition, common forms of sigma bonds are s+s, pz+pz, s+pz and dz2+dz2 . Quantum theory also indicates that molecular orbitals (MO) of identical symmetry actually mix or hybridize. As a practical consequence of this mixing of diatomic molecules, the wavefunctions s+s and pz+pz molecular orbitals become blended. The extent of this mixing depends on the relative energies of the MOs of like symmetry.
In chemistry, orbital hybridisation is the concept of mixing atomic orbitals to form new hybrid orbitals suitable for the pairing of electrons to form chemical bonds in valence bond theory. For example, in a carbon atom which forms four single bonds the valence-shell s orbital combines with three valence-shell p orbitals to form four equivalent sp3 mixtures in a tetrahedral arrangement around the carbon to bond to four different atoms. Hybrid orbitals are useful in the explanation of molecular geometry and atomic bonding properties and are symmetrically disposed in space. Usually hybrid orbitals are formed by mixing atomic orbitals of comparable energies.
In chemistry, bond order is a formal measure of the multiplicity of a covalent bond between two atoms. As introduced by Linus Pauling, bond order is defined as the difference between the numbers of electron pairs in bonding and antibonding molecular orbitals.
In chemistry, a non-covalent interaction differs from a covalent bond in that it does not involve the sharing of electrons, but rather involves more dispersed variations of electromagnetic interactions between molecules or within a molecule. The chemical energy released in the formation of non-covalent interactions is typically on the order of 1–5 kcal/mol. Non-covalent interactions can be classified into different categories, such as electrostatic, π-effects, van der Waals forces, and hydrophobic effects.

Triplet oxygen, 3O2, refers to the S = 1 electronic ground state of molecular oxygen (dioxygen). It is the most stable and common allotrope of oxygen. Molecules of triplet oxygen contain two unpaired electrons, making triplet oxygen an unusual example of a stable and commonly encountered diradical: it is more stable as a triplet than a singlet. According to molecular orbital theory, the electron configuration of triplet oxygen has two electrons occupying two π molecular orbitals (MOs) of equal energy (that is, degenerate MOs). In accordance with Hund's rules, they remain unpaired and spin-parallel and account for the paramagnetism of molecular oxygen. These half-filled orbitals are antibonding in character, reducing the overall bond order of the molecule to 2 from a maximum value of 3 (e.g., dinitrogen), which occurs when these antibonding orbitals remain fully unoccupied. The molecular term symbol for triplet oxygen is 3Σ−
g.
The 3-center 4-electron (3c–4e) bond is a model used to explain bonding in certain hypervalent molecules such as tetratomic and hexatomic interhalogen compounds, sulfur tetrafluoride, the xenon fluorides, and the bifluoride ion. It is also known as the Pimentel–Rundle three-center model after the work published by George C. Pimentel in 1951, which built on concepts developed earlier by Robert E. Rundle for electron-deficient bonding. An extended version of this model is used to describe the whole class of hypervalent molecules such as phosphorus pentafluoride and sulfur hexafluoride as well as multi-center π-bonding such as ozone and sulfur trioxide.
In theoretical chemistry, an antibonding orbital is a type of molecular orbital that weakens the chemical bond between two atoms and helps to raise the energy of the molecule relative to the separated atoms. Such an orbital has one or more nodes in the bonding region between the nuclei. The density of the electrons in the orbital is concentrated outside the bonding region and acts to pull one nucleus away from the other and tends to cause mutual repulsion between the two atoms. This is in contrast to a bonding molecular orbital, which has a lower energy than that of the separate atoms, and is responsible for chemical bonds.
In chemistry, Bent's rule describes and explains the relationship between the orbital hybridization of central atoms in molecules and the electronegativities of substituents. The rule was stated by Henry A. Bent as follows:
Atomic s character concentrates in orbitals directed toward electropositive substituents.
A molecular orbital diagram, or MO diagram, is a qualitative descriptive tool explaining chemical bonding in molecules in terms of molecular orbital theory in general and the linear combination of atomic orbitals (LCAO) method in particular. A fundamental principle of these theories is that as atoms bond to form molecules, a certain number of atomic orbitals combine to form the same number of molecular orbitals, although the electrons involved may be redistributed among the orbitals. This tool is very well suited for simple diatomic molecules such as dihydrogen, dioxygen, and carbon monoxide but becomes more complex when discussing even comparatively simple polyatomic molecules, such as methane. MO diagrams can explain why some molecules exist and others do not. They can also predict bond strength, as well as the electronic transitions that can take place.
A heavy Rydberg system consists of a weakly bound positive and negative ion orbiting their common centre of mass. Such systems share many properties with the conventional Rydberg atom and consequently are sometimes referred to as heavy Rydberg atoms. While such a system is a type of ionically bound molecule, it should not be confused with a molecular Rydberg state, which is simply a molecule with one or more highly excited electrons.
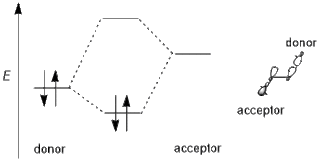
In chemistry, primarily organic and computational chemistry, a stereoelectronic effect is an effect on molecular geometry, reactivity, or physical properties due to spatial relationships in the molecules' electronic structure, in particular the interaction between atomic and/or molecular orbitals. Phrased differently, stereoelectronic effects can also be defined as the geometric constraints placed on the ground and/or transition states of molecules that arise from considerations of orbital overlap. Thus, a stereoelectronic effect explains a particular molecular property or reactivity by invoking stabilizing or destabilizing interactions that depend on the relative orientations of electrons in space.
In theoretical chemistry, the bonding orbital is used in molecular orbital (MO) theory to describe the attractive interactions between the atomic orbitals of two or more atoms in a molecule. In MO theory, electrons are portrayed to move in waves. When more than one of these waves come close together, the in-phase combination of these waves produces an interaction that leads to a species that is greatly stabilized. The result of the waves’ constructive interference causes the density of the electrons to be found within the binding region, creating a stable bond between the two species.
