Sharpless asymmetric dihydroxylation (also called the Sharpless bishydroxylation) is the chemical reaction of an alkene with osmium tetroxide in the presence of a chiralquinineligand to form a vicinaldiol. The reaction has been applied to alkenes of virtually every substitution, often high enantioselectivities are realized, with the chiral outcome controlled by the choice of dihydroquinidine (DHQD) vs dihydroquinine (DHQ) as the ligand. Asymmetric dihydroxylation reactions are also highly site selective, providing products derived from reaction of the most electron-rich double bond in the substrate.[1][2][3]
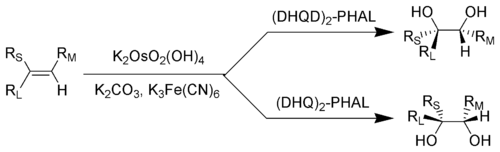
The Sharpless dihydroxylation. RL = Largest substituent; RM = Medium-sized substituent; RS = Smallest substituent
It is common practice to perform this reaction using a catalytic amount of osmium tetroxide, which after reaction is regenerated with reoxidants such as potassium ferricyanide[4][5] or N-methylmorpholine N-oxide.[6][7] This dramatically reduces the amount of the highly toxic and very expensive osmium tetroxide needed. These four reagents are commercially available premixed ("AD-mix"). The mixture containing (DHQ)2-PHAL is called AD-mix-α, and the mixture containing (DHQD)2-PHAL is called AD-mix-β.[8]
Alkene dihydroxylation by osmium tetroxide is an old and extremely useful method for the functionalization of alkenes. However, since osmium(VIII) reagents like osmium tetroxide (OsO4) are expensive and extremely toxic, it has become desirable to develop catalytic variants of this reaction. Some stoichiometric terminal oxidants that have been employed in these catalytic reactions include potassium chlorate, hydrogen peroxide (Milas hydroxylation), N-Methylmorpholine N-oxide (NMO, Upjohn dihydroxylation), tert-butyl hydroperoxide (tBHP), and potassium ferricyanide (K3Fe(CN)6). K. Barry Sharpless was the first to develop a general, reliable enantioselective alkene dihydroxylation, referred to as the Sharpless asymmetric dihydroxylation (SAD). Low levels of OsO4 are combined with a stoichiometric ferricyanide oxidant in the presence of chiral nitrogenous ligands to create an asymmetric environment around the oxidant.
Reaction mechanism
The reaction mechanism of the Sharpless dihydroxylation begins with the formation of the osmium tetroxide – ligand complex (2). A [3+2]-cycloaddition with the alkene (3) gives the cyclic intermediate 4.[9][10] Basic hydrolysis liberates the diol (5) and the reduced osmate (6). Methanesulfonamide (CH3SO2NH2) has been identified as a catalyst to accelerate this step of the catalytic cycle and if frequently used as an additive to allow non-terminal alkene substrates to react efficiently at 0 °C.[8] Finally, the stoichiometric oxidant regenerates the osmium tetroxide – ligand complex (2).
The reaction mechanism of the Sharpless dihydroxylation
The mechanism of the Sharpless asymmetric dihydroxylation has been extensively studied and a potential secondary catalytic cycle has been identified (see below).[11][12] If the osmylate ester intermediate is oxidized before it dissociates, then an osmium(VIII)-diol complex is formed which may then dihydroxylate another alkene.[13] Dihydroxylations resulting from this secondary pathway generally suffer lower enantioselectivities than those resulting from the primary pathway. A schematic showing this secondary catalytic pathway is shown below. This secondary pathway may be suppressed by using a higher molar concentration of ligand.
Catalytic cycle of the Sharpless asymmetric dihydroxylation
[2+2] vs [3+2] debate
In his original report Sharpless suggested the reaction proceeded via a [2+2] cycloaddition of OsO4 onto the alkene to give an osmaoxetane intermediate (see below).[14] This intermediate would then undergo a 1,1- migratory insertion to form an osmylate ester which after hydrolysis would give the corresponding diol. In 1989 E. J. Corey published a slightly different variant of this reaction and suggested that the reaction most likely proceeded via a [3+2] cycloaddition of OsO4 with the alkene to directly generate the osmylate ester.[15] Corey's suggestion was based on a previous computational study done by Jorgensen and Hoffmann which determined the [3+2] reaction pathway to be the lower energy pathway. In addition Corey reasoned that steric repulsions in the octahedral intermediate would disfavor the [2+2] pathway.
Osmium tetroxide dihydroxylation proposed and correct mechanism
The next ten years saw numerous publications by both Corey and Sharpless, each supporting their own version of the mechanism. While these studies were not able to distinguish between the two proposed cyclization pathways, they were successful in shedding light on the mechanism in other ways. For example, Sharpless provided evidence for the reaction proceeding via a step-wise mechanism.[16] Additionally both Sharpless and Corey showed that the active catalyst possesses a U-shaped chiral binding pocket.[17][18][19] Corey also showed that the catalyst obeys Michaelis-Menten kinetics and acts like an enzyme pocket with a pre-equilibrium.[20] In the February 1997 issue of the Journal of the American Chemical Society Sharpless published the results of a study (a Hammett analysis) which he claimed supported a [2+2] cyclization over a [3+2].[21] In the October issue of the same year, however, Sharpless also published the results of another study conducted in collaboration with Ken Houk and Singleton which provided conclusive evidence for the [3+2] mechanism.[10] Thus Sharpless was forced to concede the decade-long debate.
Catalyst structure
Allyl benzoate bound within the U-shaped binding pocket of the active dihydroquinidine catalyst, osmium tetroxide interacting with the Re face.
Crystallographic evidence has shown that the active catalyst possesses a pentacoordinate osmium species held in a U-shaped binding pocket. The nitrogenous ligand holds OsO4 in a chiral environment making approach of one side of the olefin sterically hindered while the other is not.[20]
Catalytic systems
Numerous catalytic systems and modifications have been developed for the SAD. Given below is a brief overview of the various components of the catalytic system:
Catalytic Oxidant: This is always OsO4, however certain additives can coordinate to the osmium(VIII) and modify its electronic properties. OsO4 is often generated in situ from K2OsO2(OH)4 (an Os(VI) species) due to safety concerns.
Chiral Auxiliary: This is usually some kind of cinchona alkaloid.
Stoichiometric Oxidant:
Peroxides were among the first stoichiometric oxidants to be used in this catalytic cycle; see the Milas hydroxylation. Drawbacks of peroxides include chemoselectivity issues.[13]
Trialkylammonium N-oxides, such as NMO—as in the Upjohn Reaction—and trimethylamine N-oxide.[13]
Potassium ferricyanide (K3Fe(CN)6) is the most commonly used stoichiometric oxidant for the reaction, and is the oxidant that comes in the commercially available AD-mix preparations.
Additive:
Citric acid: Osmium tetroxide is an electrophilic oxidant and as such reacts slowly with electron-deficient olefins. It has been found that the rate of oxidation of electron-deficient olefins can be accelerated by maintaining the pH of the reaction slightly acidic.[13] On the other hand, a high pH can increase the rate of oxidation of internal olefins, and also increase the enantiomeric excess (e.e.) for the oxidation of terminal olefins.[13]
Regioselectivity
In general Sharpless asymmetric dihydroxylation favors oxidation of the more electron-rich alkene (scheme 1).[22]
SAD scheme 1
In this example SAD gives the diol of the alkene closest to the (electron-withdrawing) para-methoxybenzoyl group, albeit in low yield. This is likely due to the ability of the aryl ring to interact favorably with the active site of the catalyst via π-stacking. In this manner the aryl substituent can act as a directing group.[23]
SAD scheme 2
Stereoselectivity
The diastereoselectivity of SAD is set primarily by the choice of ligand (i.e. AD-mix-α versus AD-mix-β), however factors such as pre-existing chirality in the substrate or neighboring functional groups may also play a role. In the example shown below, the para-methoxybenzoyl substituent serves primarily as a source of steric bulk to allow the catalyst to differentiate the two faces of the alkene.[23]
SAD scheme 3
It is often difficult to obtain high diastereoselectivity on cis-disubstituted alkenes when both ends of the olefin have similar steric environments.
↑ Minato, M.; Yamamoto, K.; Tsuji, J. (1990). "Osmium tetraoxide catalyzed vicinal hydroxylation of higher olefins by using hexacyanoferrate(III) ion as a cooxidant". J. Org. Chem.55 (2): 766–768. doi:10.1021/jo00289a066.
↑ VanRheenen, V.; Kelly, R. C.; Cha, D. Y. (1976). "An improved catalytic OsO4 oxidation of olefins to cis-1,2-glycols using tertiary amine oxides as the oxidant". Tetrahedron Lett.17 (23): 1973–1976. doi:10.1016/s0040-4039(00)78093-2.
1 2 Sharpless, K. B.; Amberg, Willi; Bennani, Youssef L.; etal. (1992). "The osmium-catalyzed asymmetric dihydroxylation: A new ligand class and a process improvement". J. Org. Chem.57 (10): 2768–2771. doi:10.1021/jo00036a003.
↑ Corey, E.J.; Noe, M. C.; Grogan, M. J. (1996). "Experimental test of the [3+2]- and [2+2]-cycloaddition pathways for the bis-cinchona alkaloid-OsO4 catalyzed dihydroxylation of olefins by means of kinetic isotope effects". Tetrahedron Lett.37 (28): 4899–4902. doi:10.1016/0040-4039(96)01005-2.
↑ Ogino, Y.; Chen, H.; Kwong, H.-L.; Sharpless, K. B. (1991). "On the timing of hydrolysis / reoxidation in the osmium-catalyzed asymmetric dihydroxylation of olefins using potassium ferricyanide as the reoxidant". Tetrahedron Lett. 3 (2): 3965–3968. doi:10.1016/0040-4039(91)80601-2.
↑ Wai, J. S. M.; Marko, I.; Svendsen, J. N.; Finn, M. G.; Jacobsen, E. N.; Sharpless, K. Barry (1989). "A mechanistic insight leads to a greatly improved osmium-catalyzed asymmetric dihydroxylation process". J. Am. Chem. Soc. 111 (3): 1123. Bibcode:1989JAChS.111.1123W. doi:10.1021/ja00185a050.
1 2 3 4 5 Sundermeier, U., Dobler, C., Beller, M. Recent developments in the osmium-catalyzed dihydroxylation of olefins. Modern Oxidation Methods. 2004 WILEY-VCH Verlag GmbH & Co. KGaA,Weinheim. ISBN3-527-30642-0
↑ Corey, E. J.; DaSilva Jardine, Paul; Virgil, Scott; Yuen, Po Wai; Connell, Richard D. (December 1989). "Enantioselective vicinal hydroxylation of terminal and E-1,2-disubstituted olefins by a chiral complex of osmium tetroxide. An effective controller system and a rational mechanistic model". J. Am. Chem. Soc.111 (26): 9243. Bibcode:1989JAChS.111.9243C. doi:10.1021/ja00208a025.
↑ Thomas, G.; Sharpless, K. B. ACIEE 1993, 32, 1329
↑ Corey, E. J.; Noe, Mark C. (December 1993). "Rigid and highly enantioselective catalyst for the dihydroxylation of olefins using osmium tetraoxide clarifies the origin of enantiospecificity". J. Am. Chem. Soc.26 (115): 12579. Bibcode:1993JAChS.11512579C. doi:10.1021/ja00079a045.
↑ Kolb, H. C.; Anderson, P. G.; Sharpless, K. B. (February 1994). "Toward an Understanding of the High Enantioselectivity in the Osmium-Catalyzed Asymmetric Dihydroxylation (AD). 1. Kinetics". J. Am. Chem. Soc.116 (1278): 1278. Bibcode:1994JAChS.116.1278K. doi:10.1021/ja00083a014.
↑ Corey, E. J.; Noe, Mark C.; Sarshar, Sepehr (1994). "X-ray crystallographic studies provide additional evidence that an enzyme-like binding pocket is crucial to the enantioselective dihydroxylation of olefins by OsO4-bis-cinchona alkaloid complexes". Tetrahedron Letters. 35 (18): 2861. doi:10.1016/s0040-4039(00)76644-5.
1 2 Corey, E. J.; Noe, M. C. (17 January 1996). "Kinetic Investigations Provide Additional Evidence That an Enzyme-like Binding Pocket Is Crucial for High Enantioselectivity in the Bis-Cinchona Alkaloid Catalyzed Asymmetric Dihydroxylation of Olefins". J. Am. Chem. Soc.118 (2): 319. Bibcode:1996JAChS.118..319C. doi:10.1021/ja952567z.
↑ Sharpless, K. B.; Gypser, Andreas; Ho, Pui Tong; Kolb, Hartmuth C.; Kondo, Teruyuki; Kwong, Hoi-Lun; McGrath, Dominic V.; Rubin, A. Erik; Norrby, Per-Ola; Gable, Kevin P.; Sharpless, K. Barry (1997). "Toward an Understanding of the High Enantioselectivity in the Osmium-Catalyzed Asymmetric Dihydroxylation. 4. Electronic Effects in Amine-Accelerated Osmylations". J. Am. Chem. Soc.119 (8): 1840. Bibcode:1997JAChS.119.1840N. doi:10.1021/ja961464t.
1 2 Corey, E. J.; Guzman-Perez, Angel; Noe, Mark C. (November 1995). "The application of a mechanistic model leads to the extension of the Sharpless asymmetric dihydroxylation to allylic 4-methoxybenzoates and conformationally related amine and homoallylic alcohol derivatives". J. Am. Chem. Soc.117 (44): 10805–10816. Bibcode:1995JAChS.11710805C. doi:10.1021/ja00149a003.
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.