The major advantage of this method over addition of organometallic reagents to more typical acyl compounds is that it avoids the common problem of over-addition. For these latter reactions, two equivalents of the incoming group add to form an alcohol rather than a ketone or aldehyde. This occurs even if the equivalents of nucleophile are closely controlled.
Overaddition of nucleophiles
The Weinreb–Nahm amide has since been adopted into regular use by organic chemists as a dependable method for the synthesis of ketones. These functional groups are present in a large number of natural products and can be reliably reacted to form new carbon–carbon bonds or converted into other functional groups. This method has been used in a number of syntheses, including macrosphelides A and B,[2] amphidinolide J,[3] and spirofungins A and B.[4]
Mechanism
Weinreb and Nahm originally proposed the following reaction mechanism to explain the selectivity shown in reactions of the Weinreb–Nahm amide. Their suggestion was that the tetrahedral intermediate (A below) formed as a result of nucleophilic addition by the organometallic reagent is stabilized by chelation from the methoxy group as shown.[1] This intermediate is stable only at low temperatures, requiring a low-temperature quench.
Chelation mechanism
This chelation is in contrast to the mechanism for formation of the over-addition product wherein collapse of the tetrahedral intermediate allows a second addition. The mechanistic conjecture on the part of Weinreb was immediately accepted by the academic community, but it was not until 2006 that it was confirmed by spectroscopic and kinetic analyses.[5]
Preparation
In addition to the original procedure shown above (which may have compatibility issues for sensitive substrates), Weinreb amides can be synthesized from a variety of acyl compounds. The vast majority of these procedures utilize the commercially available salt N,O-dimethylhydroxylamine hydrochloride [MeO(Me)NH•HCl], which is typically easier to handle than the free amine.[6]
Treatment of an ester or lactone with AlMe3 or AlMe2Cl affords the corresponding Weinreb amide in good yields. Alternatively, non-nucleophilic Grignard reagents such as isopropyl magnesium chloride can be used to activate the amine before addition of the ester.[7]
Finally, an aminocarbonylation reaction reported by Stephen Buchwald allows conversion of aryl halides directly into aryl Weinreb–Nahm amides.[8]
Aminocarbonylation to form Weinreb–Nahm amides
Scope
The standard conditions for the Weinreb–Nahm ketone synthesis are known to tolerate a wide variety of functional groups elsewhere in the molecule, including alpha-halogen substitution, N-protected amino acids, α-β unsaturation, silyl ethers, various lactams and lactones, sulfonates, sulfinates, and phosphonate esters.[6][7] A wide variety of nucleophiles can be used in conjunction with the amide. Lithiates and Grignard reagents are most commonly employed; examples involving aliphatic, vinyl, aryl, and alkynyl carbon nucleophiles have been reported. However, with highly basic or sterically hindered nucleophiles, elimination of the methoxide moiety to release formaldehyde can occur as a significant side reaction.[9]
Side reaction
Nonetheless, the Weinreb–Nahm amide figures prominently into many syntheses, serving as an important coupling partner for various fragments. Shown below are key steps involving Weinreb amides in the synthesis of several natural products, including members of the immunosuppressant family of macrosphelides, and the antibiotic family of spirofungins.[2][3][4]
Syntheses using Weinreb–Nahm amide
Variations
Reaction of Weinreb–Nahm amides with Wittig reagents has been performed to avoid the sometimes harsh conditions required for addition of hydride reagents or organometallic compounds. This yields an N-methyl-N-methoxy-enamine that converts to the corresponding ketone or aldehyde upon hydrolytic workup.[10]
Reaction of Weinreb–Nahm amides with Wittig reagents
Additionally, a one-pot magnesium–halogen exchange with subsequent arylation has been developed, showcasing the stability of the Weinreb–Nahm amide and providing an operationally simple method for the synthesis of aryl ketones.[11]
One-pot arylation reaction
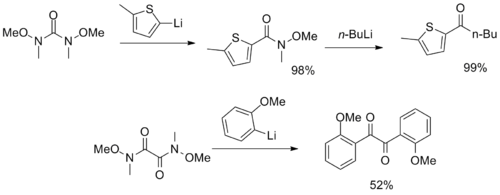
More unusual reagents with multiple Weinreb–Nahm amide functional groups have been synthesized, serving as CO2 and α-diketone synthons.[12][13]
Synthons based on Weinreb–Nahm-amides
Finally, Stephen G. Davies of Oxford has designed a chiral auxiliary that combines the functionality of the Weinreb amide with that of the Myers' pseudoephedrine auxiliary, allowing diastereoselective enolate alkylation followed by facile cleavage to the corresponding enantioenriched aldehyde or ketone.[14]
Davies' Auxiliary with Weinreb–Nahm-like functionality
↑ Martinelli, J. R.; Freckmann, D. M. M.; Buchwald, S. L. (2006), "Convenient Method for the Preparation of Weinreb Amides via Pd-Catalyzed Aminocarbonylation of Aryl Bromides at Atmospheric Pressure", Organic Letters, 8 (21): 4843–4846, doi:10.1021/ol061902t, PMID17020317
↑ Whipple, W. L.; Reich, H. J. (1991), "Use of N,N'-dimethoxy-N,N'-dimethylurea as a carbonyl dication equivalent in organometallic addition reactions. Synthesis of unsymmetrical ketones", The Journal of Organic Chemistry, 56 (8): 2911–2912, doi:10.1021/jo00008a057
↑ Sibi, M. P.; Sharma, R.; Paulson, K. L. (1992), "N,N′-Dimethoxy-N,N -Dimethylethanediamide: A Useful α-Oxo-N-Methoxy-N-Methylamide and 1,2-Diketone Synthon", Tetrahedron Letters, 33 (15): 1941–1944, doi:10.1016/0040-4039(92)88108-h
↑ Davies, S. G.; Goodwin, C. J.; Hepworth, D.; Roberts, P. M.; Thomson, J. E. (2010), "On the Origins of Diastereoselectivity in the Alkylation of Enolates Derived from N-1-(1'-Naphthyl)ethyl-O-tert-butylhydroxamates: Chiral Weinreb Amide Equivalents", The Journal of Organic Chemistry, 75 (4): 1214–1227, doi:10.1021/jo902499s, PMID20095549
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.