Iodolactonization (or, more generally, halolactonization) is an organic reaction that forms a ring (the lactone) by the addition of an oxygen and iodine across a carbon-carbon double bond. It is an intramolecular variant of the halohydrin synthesis reaction. The reaction was first reported by M. J. Bougalt in 1904 and has since become one of the most effective ways to synthesize lactones.[1] Strengths of the reaction include the mild conditions and incorporation of the versatile iodine atom into the product.
Bougalt's report of iodolactonization represented the first example of a reliable lactonization that could be used in many different systems. Bromolactonization was actually developed in the twenty years prior to Bougalt’s publication of iodolactonization.[1] However, bromolactonization is much less commonly used because the simple electrophilic addition of bromine to an alkene, seen below, can compete with the bromolactonization reaction and reduce the yield of the desired lactone.[5]
Bromolactonization
Chlorolactonization methods first appeared in the 1950s[1] but are even less commonly employed than bromolactonization. The use of elemental chlorine is procedurally difficult because it is a gas at room temperature, and the electrophilic addition product can be rapidly produced as in bromolactonization.[6]
Mechanism
The reaction mechanism involves the formation of a positively charged halonium ion in a molecule that also contains a carboxylic acid (or other functional group that is a precursor to it). The oxygen of the carboxyl acts as a nucleophile, attacking to open the halonium ring and instead form a lactone ring. The reaction is usually performed under mildly basic conditions to increase the nucleophilicity of the carboxyl group.
Iodolactonization
Scope
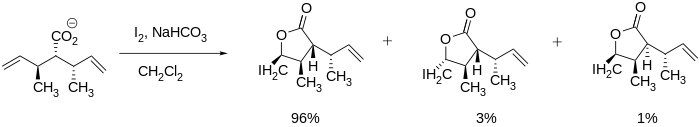
The iodolactonization reaction includes a number of nuances that affect product formation including regioselectivity, ring size preference, and thermodynamic and kinetic control. In terms of regioselectivity, iodolactonization preferentially occurs at the most hindered carbon atom adjacent to the iodonium cation. This is due to the fact that the more substituted carbon is better able to maintain a partial positive charge and is thus more electrophilic and susceptible to nucleophilic attack. When multiple double bonds in a molecule are equally reactive, conformational preferences dominate. However, when one double bond is more reactive, that reactivity always dominates regardless of conformational preference.[7]
IodolactonizationRegioselectivity
Both five- and six-membered rings could be formed in the iodolactonization shown below, but the five-membered ring is formed preferentially as predicted by Baldwin's rules for ring closure.[8] According to the rules, 5-exo-tet ring closures are favored while 6-endo-tet ring closures are disfavored.[9] The regioselectivity of each iodolactonization can be predicted and explained using Baldwin's rules.
Iodolactonization
Stereoselective iodolactonizations have been seen in literature and can be very useful in synthesizing large molecules such as the aforementioned vernopelin and vernomenin because the lactone can be formed while maintaining other stereocenters. The ring closure can even be driven by stereocenters adjacent to the carbon-carbon multiple bond as shown below.[7]
Iodolactonization
Even in systems without existing stereocenters, Bartlett and coworkers found that stereoselectivity was achievable. They were able to synthesize the cis and trans five membered lactones by adjusting reactions conditions such as temperature and reaction time. The trans product was formed under thermodynamic conditions (e.g. a long reaction time) while the cis product was formed under kinetic conditions (e.g. a relatively shorter reaction time).[10]
Iodolactonization
Applications
Iodolactonization has been used in the synthesis of many biologically important products such as the tumor growth inhibitors vernolepin and vernomenin, the pancreatic lipase inhibitor vibralactone, and prostaglandins, a lipid found in animals. The following total syntheses all use iodolactonization as a key step in obtaining the desired product.
In 1977, Samuel Danishefsky and coworkers were able to synthesize the tumor growth inhibitors dl-vernolepin and dl-vernomenin via a multistep process in which a lactonization was employed.[2] This synthesis demonstrates the use of iodolactonization to preferentially form a five-membered ring over a four- or six-membered ring, as expected from Baldwin's rules.
Danishefsky iodolactonization
In 2006, Zhou and coworkers synthesized another natural product, vibralactone, in which the key step was the formation of a lactone.[3] The stereoselectivity of the iodolactonization sets a critical stereochemical configuration for the target compound.
Iodolactonization
In 1969, Corey and coworkers synthesized prostaglandin E2 using an iodolactone intermediate.[4] Again, the stereoselectivity of the iodolactonization plays an integral role in product formation.
1 2 3 Dowle, M. D.; Davies, D. I. (1979). "Synthesis and synthetic utility of halolactones". Chemical Society Reviews. 8 (2): 171. doi:10.1039/CS9790800171.
1 2 Danishefsky, S.; Schuda, P. F.; Kitahara, T.; Etheredge, S. J. (1977). "The total synthesis of dl-vernolepin and dl-vernomenin". Journal of the American Chemical Society. 99 (18): 6066. doi:10.1021/ja00460a038.
1 2 Corey, E. J.; Weinshenker, N. M.; Schaaf, T. K.; Huber, W. (1969). "Stereo-controlled synthesis of dl-prostaglandins F2α And E2". Journal of the American Chemical Society. 91 (20): 5675–5677. doi:10.1021/ja01048a062. PMID5808505.
↑ Brown, R. S. (1997). "Investigation of the Early Steps in Electrophilic Bromination through the Study of the Reaction with Sterically Encumbered Olefins". Accounts of Chemical Research. 30 (3): 131–137. doi:10.1021/ar960088e.
↑ Garratt, D. G.; Ryan, M. D.; Beaulieu, P. L. (1980). "Additions of Group 6A and 7A electrophilic reagents to dimethyl endo,endo-bicyclo[2.2.2]oct-5-ene-2,3-dicarboxylate: Competitive formation of γ- and δ-lactones". The Journal of Organic Chemistry. 45 (5): 839. doi:10.1021/jo01293a016.
1 2 Kurth, M. J.; Brown, E. G.; Lewis, E. J.; McKew, J. C. (1988). "Regioselectivity in the iodolactonization of 1,6-heptadien-4-carboxylic acid derivatives". Tetrahedron Letters. 29 (13): 1517. doi:10.1016/S0040-4039(00)80340-8.
↑ Baldwin, Jack E. (1976). "Rules for ring closure". Journal of the Chemical Society, Chemical Communications (18): 734. doi:10.1039/c39760000734. ISSN0022-4936.
↑ Bartlett, P. A.; Myerson, J. (1978). "Stereoselective epoxidation of acyclic olefinic carboxylic acids via iodolactonization". Journal of the American Chemical Society. 100 (12): 3950. doi:10.1021/ja00480a061.
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.