These organostannanes are also stable to both air and moisture, and many of these reagents either are commercially available or can be synthesized from literature precedent. However, these tin reagents tend to be highly toxic. X is typically a halide, such as Cl, Br, or I, yet pseudohalides such as triflates and sulfonates and phosphates can also be used.[4][5] Several reviews have been published.[6][2][7][8][9][10][11][12][13][14][15][excessive citations]
In 1977, Migita published further work on the coupling of allyl-tin reagents with both aryl (C) and acyl (D) halides. The greater ability of allyl groups to migrate to the palladium catalyst allowed the reactions to be performed at lower temperatures. Yields for aryl halides ranged from 4% to 100%, and for acyl halides from 27% to 86%.[18][19] Reflecting the early contributions of Migita and Kosugi, the Stille reaction is sometimes called the Migita–Kosugi–Stille coupling.
First reactions of organotin reagents
John Kenneth Stille subsequently reported the coupling of a variety of alkyl tin reagents in 1978 with numerous aryl and acyl halides under mild reaction conditions with much better yields (76%–99%).[18][20] Stille continued his work in the 1980s on the synthesis of a multitude of ketones using this broad and mild process and elucidated a mechanism for this transformation.[21][22]
First reactions of organotin reagents
By the mid-1980s, over 65 papers on the topic of coupling reactions involving tin had been published, continuing to explore the substrate scope of this reaction. While initial research in the field focused on the coupling of alkyl groups, most future work involved the much more synthetically useful coupling of vinyl, alkenyl, aryl, and allyl organostannanes to halides. Due to these organotin reagent's stability to air and their ease of synthesis, the Stille reaction became common in organic synthesis.[8]
However, the detailed mechanism of the Stille coupling is extremely complex and can occur via numerous reaction pathways. Like other palladium-catalyzed coupling reactions, the active palladium catalyst is believed to be a 14-electron Pd(0) complex, which can be generated in a variety of ways. Use of an 18- or 16- electron Pd(0) source Pd(PPh3)4, Pd(dba)2 can undergo ligand dissociation to form the active species. Second, phosphines can be added to ligandless palladium(0). Finally, as pictured, reduction of a Pd(II) source (8) (Pd(OAc)2, PdCl2(MeCN)2, PdCl2(PPh3)2, BnPdCl(PPh3)2, etc.) by added phosphine ligands or organotin reagents is also common[6]
Oxidative addition
Oxidative addition to the 14-electron Pd(0) complex is proposed. This process gives a 16-electron Pd(II) species. It has been suggested that anionic ligands, such as OAc, accelerate this step by the formation of [Pd(OAc)(PR3)n]−, making the palladium species more nucleophillic.[11][25] In some cases, especially when an sp3-hybridizedorganohalide is used, an SN2 type mechanism tends to prevail, yet this is not as commonly seen in the literature.[11][25] However, despite normally forming a cis-intermediate after a concerted oxidative addition, this product is in rapid equilibrium with its trans-isomer.[26][27]
Cis/Trans Isomerization
There are multiple reasons why isomerization is favored here. First, a bulky ligand set is usually used in these processes, such as phosphines, and it is highly unfavorable for them to adopt a cis orientation relative to each other, resulting in isomerization to the more favorable trans product.[26][27] An alternative explanation for this phenomenon, dubbed antisymbiosis or transphobia, is by invocation of the sdn model.[24][28] Under this theory, palladium is a hypervalent species. Hence R1 and the trans ligand, being trans to each other, will compete with one palladium orbital for bonding. This 4-electron 3-center bond is weakest when two strong donating groups are present, which heavily compete for the palladium orbital. Relative to any ligand normally used, the C-donor R1 ligand has a much higher trans effect. This trans influence is a measure of how competitive ligands trans to each other will compete for palladium's orbital. The usual ligand set, phosphines, and C-donors (R1) are both soft ligands, meaning that they will form strong bonds to palladium, and heavily compete with each other for bonding.[29][30] Since halides or pseudohalides are significantly more electronegative, their bonding with palladium will be highly polarized, with most of the electron density on the X group, making them low trans effect ligands. Hence, it will be highly favorable for R1 to be trans to X, since the R1 group will be able to form a stronger bond to the palladium.[24][28][30]
sd^n model for cis/trans isomers
Transmetallation
The transmetallation of the trans intermediate from the oxidative addition step is believed to proceed via a variety of mechanisms depending on the substrates and conditions. The most common type of transmetallation for the Stille coupling involves an associative mechanism. This pathway implies that the organostannane, normally a tin atom bonded to an allyl, alkenyl, or aryl group, can coordinate to the palladium via one of these double bonds. This produces a fleeting pentavalent, 18-electron species, which can then undergo ligand detachment to form a square planar complex again. Despite the organostannane being coordinated to the palladium through the R2 group, R2 must be formally transferred to the palladium (the R2-Sn bond must be broken), and the X group must leave with the tin, completing the transmetalation. This is believed to occur through two mechanisms.[31]
First, when the organostannane initially adds to the trans metal complex, the X group can coordinate to the tin, in addition to the palladium, producing a cyclic transition state. Breakdown of this adduct results in the loss of R3Sn-X and a trivalent palladium complex with R1 and R2 present in a cis relationship. Another commonly seen mechanism involves the same initial addition of the organostannane to the trans palladium complex as seen above; however, in this case, the X group does not coordinate to the tin, producing an open transition state. After the α-carbon relative to tin attacks the palladium, the tin complex will leave with a net positive charge. In the scheme below, please note that the double bond coordinating to tin denotes R2, so any alkenyl, allyl, or aryl group. Furthermore, the X group can dissociate at any time during the mechanism and bind to the Sn+ complex at the end. Density functional theory calculations predict that an open mechanism will prevail if the 2 ligands remain attached to the palladium and the X group leaves, while the cyclic mechanism is more probable if a ligand dissociates prior to the transmetalation. Hence, good leaving groups such as triflates in polar solvents favor the cyclic transition state, while bulky phosphine ligands will favor the open transition state.[31]
The two mechanisms, cyclic and open, of transmetallation in the Stille reaction
A less common pathway for transmetalation is through a dissociative or solvent assisted mechanism. Here, a ligand from the tetravalent palladium species dissociates, and a coordinating solvent can add onto the palladium. When the solvent detaches, to form a 14-electron trivalent intermediate, the organostannane can add to the palladium, undergoing an open or cyclic type process as above.[31]
Reductive elimination step
In order for R1-R2 to reductively eliminate, these groups must occupy mutually cis coordination sites. Any trans-adducts must therefore isomerize to the cis intermediate or the coupling will be frustrated. A variety of mechanisms exist for reductive elimination and these are usually considered to be concerted.[11][32][33]
First, the 16-electron tetravalent intermediate from the transmetalation step can undergo unassisted reductive elimination from a square planar complex. This reaction occurs in two steps: first, the reductive elimination is followed by coordination of the newly formed sigma bond between R1 and R2 to the metal, with ultimate dissociation yielding the coupled product.[11][32][33]
Concerted reductive elimination for the Stille reaction
The previous process, however, is sometimes slow and can be greatly accelerated by dissociation of a ligand to yield a 14-electron T shaped intermediate. This intermediate can then rearrange to form a Y-shaped adduct, which can undergo faster reductive elimination.[11][32][33]
Dissociative reductive elimination for the Stille reaction
Finally, an extra ligand can associate to the palladium to form an 18-electron trigonal bipyramidal structure, with R1 and R2 cis to each other in equatorial positions. The geometry of this intermediate makes it similar to the Y-shaped above.[11][32][33]
Associative reductive elimination for the Stille reaction
The presence of bulky ligands can also increase the rate of elimination. Ligands such as phosphines with large bite angles cause steric repulsion between L and R1 and R2, resulting in the angle between L and the R groups to increase and the angle between R1 and R2 to hence decrease, allowing for quicker reductive elimination.[11][24]
Cis-reductive elimination in the Stille reaction
Kinetics
The rate at which organostannanes transmetalate with palladium catalysts is shown below. Sp2-hybridized carbon groups attached to tin are the most commonly used coupling partners, and sp3-hybridized carbons require harsher conditions and terminal alkynes may be coupled via a C-H bond through the Sonogashira reaction.
Relative rates of the Stille reaction
As the organic tin compound, a trimethylstannyl or tributylstannyl compound is normally used. Although trimethylstannyl compounds show higher reactivity compared with tributylstannyl compounds and have much simpler 1H-NMR spectra, the toxicity of the former is much larger.[34]
Optimizing which ligands are best at carrying out the reaction with high yield and turnover rate can be difficult. This is because the oxidative addition requires an electron rich metal, hence favoring electron donating ligands. However, an electron deficient metal is more favorable for the transmetalation and reductive elimination steps, making electron withdrawing ligands the best here. Therefore, the optimal ligand set heavily depends on the individual substrates and conditions used. These can change the rate determining step, as well as the mechanism for the transmetalation step.[35]
Normally, ligands of intermediate donicity, such as phosphines, are utilized. Rate enhancements can be seen when moderately electron-poor ligands, such as tri-2-furylphosphine or triphenylarsenine are used. Likewise, ligands of high donor number can slow down or inhibit coupling reactions.[35][36]
These observations imply that normally, the rate-determining step for the Stille reaction is transmetalation.[36]
Lithium chloride has been found to be a powerful rate accelerant in cases where the X group dissociates from palladium (i.e. the open mechanism). The chloride ion is believed to either displace the X group on the palladium making the catalyst more active for transmetalation or by coordination to the Pd(0) adduct to accelerate the oxidative addition. Also, LiCl salt enhances the polarity of the solvent, making it easier for this normally anionic ligand (–Cl, –Br, –OTf, etc.) to leave. This additive is necessary when a solvent like THF is used; however, utilization of a more polar solvent, such as NMP, can replace the need for this salt additive. However, when the coupling's transmetalation step proceeds via the cyclic mechanism, addition of lithium chloride can actually decrease the rate. As in the cyclic mechanism, a neutral ligand, such as phosphine, must dissociate instead of the anionic X group.[10][41]
The most common side reactivity associated with the Stille reaction is homocoupling of the stannane reagents to form an R2-R2dimer. It is believed to proceed through two possible mechanisms. First, reaction of two equivalents of organostannane with the Pd(II) precatalyst will yield the homocoupled product after reductive elimination. Second, the Pd(0) catalyst can undergo a radical process to yield the dimer. The organostannane reagent used is traditionally tetravalent at tin, normally consisting of the sp2-hybridized group to be transferred and three "non-transferable" alkyl groups. As seen above, alkyl groups are normally the slowest at migrating onto the palladium catalyst.[10]
Homocoupling and transfer of "inert" ligands
It has also been found that at temperatures as low as 50°C, aryl groups on both palladium and a coordinatedphosphine can exchange. While normally not detected, they can be a potential minor product in many cases.[10]
Numerous other side reactions can occur, and these include E/Z isomerization, which can potentially be a problem when an alkenylstannane is utilized. The mechanism of this transformation is currently unknown. Normally, organostannanes are quite stable to hydrolysis, yet when very electron-rich aryl stannanes are used, this can become a significant side reaction.[10]
Scope
Electrophile
Vinyl halides are common coupling partners in the Stille reaction, and reactions of this type are found in numerous natural producttotal syntheses. Normally, vinyl iodides and bromides are used. Vinyl chlorides are insufficiently reactive toward oxidative addition to Pd(0). Iodides are normally preferred: they will typically react faster and under milder conditions than will bromides. This difference is demonstrated below by the selective coupling of a vinyl iodide in the presence of a vinyl bromide.[10]
Vinyl Iodide reacts faster than vinyl bromide
Normally, the stereochemistry of the alkene is retained throughout the reaction, except under harsh reaction conditions. A variety of alkenes may be used, and these include both α- and β-halo-α,β unsaturated ketones, esters, and sulfoxides (which normally need a copper (I) additive to proceed), and more (see example below).[42] Vinyl triflates are also sometimes used. Some reactions require the addition of LiCl and others are slowed down, implying that two mechanistic pathways are present.[10]
Addition to an alpha, beta unsaturated alkene
Another class of common electrophiles are aryl and heterocyclic halides, especially bromides and iodides.[10] Aryl triflates and sulfonates are also couple to a wide variety of organostannane reagents. Triflates tend to react comparably to bromides in the Stille reaction.[10]
Acyl chlorides are also used as coupling partners and can be used with a large range of organostannane, even alkyl-tin reagents, to produce ketones (see example below).[43] However, it is sometimes difficult to introduce acyl chloride functional groups into large molecules with sensitive functional groups. An alternative developed to this process is the Stille-carbonylative cross-coupling reaction, which introduces the carbonyl group via carbon monoxide insertion.[10]
Acyl chlorides can be used as well
Allylic, benzylic, and propargylic halides can also be coupled. While commonly employed, allylic halides proceed via an η3 transition state, allowing for coupling with the organostannane at either the α or γ position, occurring predominantly at the least substituted carbon (see example below).[44] Alkenyl epoxides (adjacent epoxides and alkenes) can also undergo this same coupling through an η3transition state as, opening the epoxide to an alcohol. While allylic and benzylic acetates are commonly used, propargylic acetates are unreactive with organostannanes.[10]
allylic bromides will form an heta-3 complex.
Stannane
Organostannane reagents are common. Several are commercially available.[45] Stannane reagents can be synthesized by the reaction of a Grignard or organolithium reagent with trialkyltin chlorides. For example, vinyltributyltin is prepared by the reaction of vinylmagnesium bromide with tributyltin chloride.[46]Hydrostannylation of alkynes or alkenes provides many derivatives. Organotin reagents are air and moisture stable. Some reactions can even take place in water.[47] They can be purified by chromatography. They are tolerant to most functional groups. Some organotin compounds are heavily toxic, especially trimethylstannyl derivatives.[10]
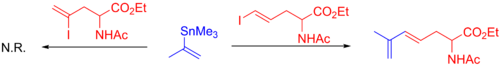
The use of vinylstannane, or alkenylstannane reagents is widespread.[10] In regards to limitations, both very bulky stannane reagents and stannanes with substitution on the α-carbon tend to react sluggishly or require optimization. For example, in the case below, the α-substituted vinylstannane only reacts with a terminal iodide due to steric hindrance.[48]
Stannae 1
Arylstannane reagents are also common and both electron donating and electron withdrawing groups actually increase the rate of the transmetalation. This again implies that two mechanisms of transmetalation can occur. The only limitation to these reagents are substituents at the ortho-position as small as methyl groups can decrease the rate of reaction. A wide variety of heterocycles (see Electrophile section) can also be used as coupling partners (see example with a thiazole ring below).[10][49]
Regioselective coupling of a heterocyclic-stannae with an aryl bromideCoupling of stannane to acyl chloride
Alkynylstannanes, the most reactive of stannanes, have also been used in Stille couplings. They are not usually needed as terminal alkynes can couple directly to palladium catalysts through their C-H bond via Sonogashira coupling. Allylstannanes have been reported to have worked, yet difficulties arise, like with allylic halides, with the difficulty in control regioselectivity for α and γ addition. Distannane and acyl stannane reagents have also been used in Stille couplings.[10]
Natural product syntheses and other reactions
The total synthesis of quadrigemine C involves a double Stille cross metathesis reaction.[6][50] The complex organostannane is coupled onto two aryl iodide groups. After a double Heck cyclization, the product is achieved.
Total Synthesis of Quadrigemine C
The synthesis of ansamycinantibiotic (+)-mycotrienol makes use of a late stage tandem Stille type macrocycle coupling. Here, the organostannane has two terminal tributyl tin groups attacked to an alkene. This organostannane "stitches" the two ends of the linear starting material into a macrocycle, adding the missing two methylene units in the process. After oxidation of the aromatic core with ceric ammonium nitrate (CAN) and deprotection with hydrofluoric acid yields the natural product in 54% yield for the 3 steps.[6][51]
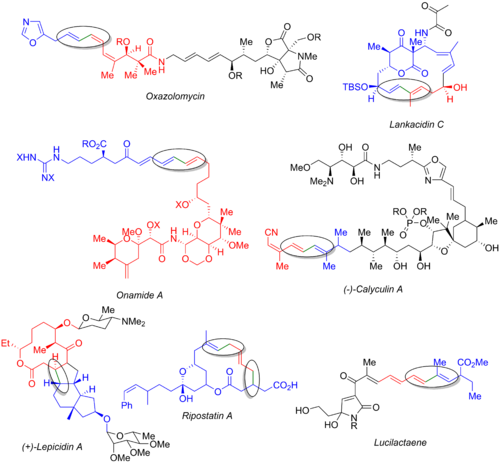
Numerous other total syntheses utilize the Stille reaction, including those of oxazolomycin,[53] lankacidin C,[54] onamide A,[55] calyculin A,[56] lepicidin A,[57] ripostatin A,[58] and lucilactaene.[6][59] The image below displays the final natural product, the organohalide (blue), the organostannane (red), and the bond being formed (green and circled). From these examples, it is clear that the Stille reaction can be used both at the early stages of the synthesis (oxazolomycin and calyculin A), at the end of a convergent route (onamide A, lankacidin C, ripostatin A), or in the middle (lepicidin A and lucilactaene). The synthesis of ripostatin A features two concurrent Stille couplings followed by a ring-closing metathesis. The synthesis of lucilactaene features a middle subunit, having a borane on one side and a stannane on the other, allowing for a Stille reaction followed by a subsequent Suzuki coupling.
A variety of total syntheses which make use of the Stille reaction
The Stille reaction has been used in the synthesis of a variety of polymers.[60][61][62]
Variations
In addition to performing the reaction in a variety of organic solvents, conditions have been devised which allow for a broad range of Stille couplings in aqueous solvent.[14]
A Stille reaction variation: coupling of phenyliodide and tetramethyltin
Stille–carbonylative cross-coupling
A common alteration to the Stille coupling is the incorporation of a carbonyl group between R1 and R2, serving as an efficient method to form ketones. This process is extremely similar to the initial exploration by Migita and Stille (see History) of coupling organostannane to acyl chlorides. However, these moieties are not always readily available and can be difficult to form, especially in the presence of sensitive functional groups. Furthermore, controlling their high reactivity can be challenging. The Stille-carbonylative cross-coupling employs the same conditions as the Stille coupling, except with an atmosphere of carbon monoxide (CO) being used. The CO can coordinate to the palladium catalyst (9) after initial oxidative addition, followed by CO insertion into the Pd-R1 bond (10), resulting in subsequent reductive elimination to the ketone (12). The transmetalation step is normally the rate-determining step.[6]
Catalytic cycle of the Stille-carbonylative cross-coupling
Giorgio Ortar et al. explored how the Stille-carbonylative cross-coupling could be used to synthesize benzophenone phosphores. These were embedded into 4-benzoyl-L-phenylalanine peptides and used for their photoaffinity labelling properties to explore various peptide-protein interactions.[6][68]
Synthesis of phosphores
Louis Hegedus' 16-step racemictotal synthesis of Jatraphone involved a Stille-carbonylative cross-coupling as its final step to form the 11-membered macrocycle. Instead of a halide, a vinyl triflate is used there as the coupling partner.[6][69]
Total synthesis of Jatraphone
Stille–Kelly coupling
Using the seminal publication by Eaborn in 1976, which forms arylstannanes from arylhalides and distannanes, T. Ross Kelly applied this process to the intramolecular coupling of arylhalides. This tandem stannylation/aryl halide coupling was used for the syntheses of a variety of dihydrophenanthrenes. Most of the internal rings formed are limited to 5 or 6 members, however some cases of macrocyclization have been reported. Unlike a normal Stille coupling, chlorine does not work as a halogen, possibly due to its lower reactivity in the halogen sequence (its shorter bond length and stronger bond dissociation energy makes it more difficult to break via oxidative addition). Starting in the middle of the scheme below and going clockwise, the palladium catalyst (1) oxidatively adds to the most reactive C-X bond (13) to form 14, followed by transmetalation with distannane (15) to yield 16 and reductive elimination to yield an arylstannane (18). The regenerated palladium catalyst (1) can oxidative add to the second C-X bond of 18 to form 19, followed by intramolecular transmetalation to yield 20, followed by reductive elimination to yield the coupled product (22).[6]
Catalytic cycle of the Stille-Kelly reaction
Jie Jack Lie et al. made use of the Stille-Kelly coupling in their synthesis of a variety of benzo[4,5]furopyridines ring systems. They invoke a three-step process, involving a Buchwald-Hartwig amination, another palladium-catalyzed coupling reaction, followed by an intramolecular Stille-Kelly coupling. Note that the aryl-iodide bond will oxidatively add to the palladium faster than either of the aryl-bromide bonds.[6][70]
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.





























![Synthesis of benzo[4,5]furopyridines Benzofuropyridines.png](http://upload.wikimedia.org/wikipedia/commons/thumb/2/25/Benzofuropyridines.png/500px-Benzofuropyridines.png)