
A chemical reaction is a process that leads to the chemical transformation of one set of chemical substances to another. Classically, chemical reactions encompass changes that only involve the positions of electrons in the forming and breaking of chemical bonds between atoms, with no change to the nuclei, and can often be described by a chemical equation. Nuclear chemistry is a sub-discipline of chemistry that involves the chemical reactions of unstable and radioactive elements where both electronic and nuclear changes can occur.
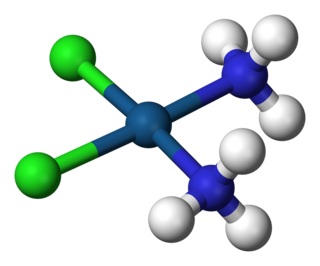
A coordination complex consists of a central atom or ion, which is usually metallic and is called the coordination centre, and a surrounding array of bound molecules or ions, that are in turn known as ligands or complexing agents. Many metal-containing compounds, especially those of transition metals, are coordination complexes. A coordination complex whose centre is a metal atom is called a metal complex.
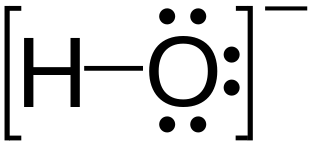
Hydroxide is a diatomic anion with chemical formula OH−. It consists of an oxygen and hydrogen atom held together by a covalent bond, and carries a negative electric charge. It is an important but usually minor constituent of water. It functions as a base, a ligand, a nucleophile, and a catalyst. The hydroxide ion forms salts, some of which dissociate in aqueous solution, liberating solvated hydroxide ions. Sodium hydroxide is a multi-million-ton per annum commodity chemical. A hydroxide attached to a strongly electropositive center may itself ionize, liberating a hydrogen cation (H+), making the parent compound an acid.
Inorganic chemistry deals with the synthesis and behavior of inorganic and organometallic compounds. This field covers all chemical compounds except the myriad organic compounds, which are the subjects of organic chemistry. The distinction between the two disciplines is far from absolute, as there is much overlap in the subdiscipline of organometallic chemistry. It has applications in every aspect of the chemical industry, including catalysis, materials science, pigments, surfactants, coatings, medications, fuels, and agriculture.
In coordination chemistry, a ligand is an ion or molecule that binds to a central metal atom to form a coordination complex. The bonding with the metal generally involves formal donation of one or more of the ligand's electron pairs. The nature of metal–ligand bonding can range from covalent to ionic. Furthermore, the metal–ligand bond order can range from one to three. Ligands are viewed as Lewis bases, although rare cases are known to involve Lewis acidic "ligands".
The Stille reaction is a chemical reaction widely used in organic synthesis. The involves the coupling of two organic groups, one of which is carried as an organotin compound. A variety organic electrophiles provide the other coupling partner. The Stille reaction is one of many palladium-catalyzed coupling reactions.

The isolobal principle is a strategy used in organometallic chemistry to relate the structure of organic and inorganic molecular fragments in order to predict bonding properties of organometallic compounds. Roald Hoffmann described molecular fragments as isolobal "if the number, symmetry properties, approximate energy and shape of the frontier orbitals and the number of electrons in them are similar – not identical, but similar." One can predict the bonding and reactivity of a lesser-known species from that of a better-known species if the two molecular fragments have similar frontier orbitals, the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO). Isolobal compounds are analogues to isoelectronic compounds that share the same number of valence electrons and structure. A graphic representation of isolobal structures, with the isolobal pairs connected through a double-headed arrow with half an orbital below, is found in Figure 1.
Reductive elimination is an elementary step in organometallic chemistry in which the oxidation state of the metal center decreases while forming a new covalent bond between two ligands. It is the microscopic reverse of oxidative addition, and is often the product-forming step in many catalytic processes. Since oxidative addition and reductive elimination are reverse reactions, the same mechanisms apply for both processes, and the product equilibrium depends on the thermodynamics of both directions.
Oxidative addition and reductive elimination are two important and related classes of reactions in organometallic chemistry. Oxidative addition is a process that increases both the oxidation state and coordination number of a metal centre. Oxidative addition is often a step in catalytic cycles, in conjunction with its reverse reaction, reductive elimination.

In coordination chemistry, the first coordination sphere refers to the array of molecules and ions directly attached to the central metal atom. The second coordination sphere consists of molecules and ions that attached in various ways to the first coordination sphere.

Hapticity is the coordination of a ligand to a metal center via an uninterrupted and contiguous series of atoms. The hapticity of a ligand is described with the Greek letter η ('eta'). For example, η2 describes a ligand that coordinates through 2 contiguous atoms. In general the η-notation only applies when multiple atoms are coordinated. In addition, if the ligand coordinates through multiple atoms that are not contiguous then this is considered denticity, and the κ-notation is used once again. When naming complexes care should be taken not to confuse η with μ ('mu'), which relates to bridging ligands.
The 18-electron rule is a rule used primarily for predicting and rationalizing formulas for stable metal complexes, especially organometallic compounds. The rule is based on the fact that the valence shells of transition metals consist of nine valence orbitals, which collectively can accommodate 18 electrons as either bonding or nonbonding electron pairs. This means that the combination of these nine atomic orbitals with ligand orbitals creates nine molecular orbitals that are either metal-ligand bonding or non-bonding. When a metal complex has 18 valence electrons, it is said to have achieved the same electron configuration as the noble gas in the period. The rule and its exceptions are similar to the application of the octet rule to main group elements. The rule is not helpful for complexes of metals that are not transition metals, and interesting or useful transition metal complexes will violate the rule because of the consequences deviating from the rule bears on reactivity. The rule was first proposed by American chemist Irving Langmuir in 1921.
The SN1cB (conjugate base) mechanism describes the pathway by which many metal amine complexes undergo substitution, that is ligand exchange. Typically, the reaction entails reaction of a polyamino metal halide with aqueous base to give the corresponding polyamine metal hydroxide:
A migratory insertion is a type of reaction in organometallic chemistry wherein two ligands on a metal complex combine. It is a subset of reactions that very closely resembles the insertion reactions, and both are differentiated by the mechanism that leads to the resulting stereochemistry of the products. However, often the two are used interchangeably because the mechanism is sometimes unknown. Therefore, migratory insertion reactions or insertion reactions, for short, are defined not by the mechanism but by the overall regiochemistry wherein one chemical entity interposes itself into an existing bond of typically a second chemical entity e.g.:
In organometallic chemistry, a transition metal indenyl complex is a coordination compound that contains one or more indenyl ligands. The indenyl ligand is formally the anion derived from deprotonation of indene. The η5-indenyl ligand is related to the η5cyclopentadienyl anion (Cp), thus indenyl analogues of many cyclopentadienyl complexes are known. Indenyl ligands lack the 5-fold symmetry of Cp, so they exhibit more complicated geometries. Furthermore, some indenyl complexes also exist with only η3-bonding mode. The η5- and η3-bonding modes sometimes interconvert.
A stability constant is an equilibrium constant for the formation of a complex in solution. It is a measure of the strength of the interaction between the reagents that come together to form the complex. There are two main kinds of complex: compounds formed by the interaction of a metal ion with a ligand and supramolecular complexes, such as host–guest complexes and complexes of anions. The stability constant(s) provide the information required to calculate the concentration(s) of the complex(es) in solution. There are many areas of application in chemistry, biology and medicine.
Dissociative substitution describes a pathway by which compounds interchange ligands. The term is typically applied to coordination and organometallic complexes, but resembles the Sn1 mechanism in organic chemistry. This pathway can be well described by the cis effect, or the labilization of CO ligands in the cis position. The opposite pathway is associative substitution, being analogous to Sn2 pathway. Intermediate pathways exist between the pure dissociative and pure associative pathways, these are called interchange mechanisms.
Metal aquo complexes are coordination compounds containing metal ions with only water as a ligand. These complexes are the predominant species in aqueous solutions of many metal salts, such as metal nitrates, sulfates, and perchlorates. They have the general stoichiometry [M(H2O)n]z+. Their behavior underpins many aspects of environmental, biological, and industrial chemistry. This article focuses on complexes where water is the only ligand ("homoleptic aquo complexes"), but of course many complexes are known to consist of a mix of aquo and other ligands.
In inorganic chemistry, the cis effect is defined as the labilization (making unstable) of CO ligands that are cis to other ligands. CO is a well-known strong pi-accepting ligand in organometallic chemistry that will labilize in the cis position when adjacent to ligands due to steric and electronic effects. The system most often studied for the cis effect is an octahedral complex M(CO)5X where X is the ligand that will labilize a CO ligand cis to it. Unlike trans effect, where this property is most often observed in 4-coordinate square planar complexes, the cis effect is observed in 6-coordinate octahedral transition metal complexes. It has been determined that ligands that are weak sigma donors and non-pi acceptors seem to have the strongest cis-labilizing effects. Therefore, the cis effect has the opposite trend of the trans-effect, which effectively labilizes ligands that are trans to strong pi accepting and sigma donating ligands.

Half sandwich compounds are organometallic complexes that feature a cyclic polyhapto ligand bound to an MLn center, where L is an unidentate ligand. Thousands of such complexes are known. Well-known examples include cyclobutadieneiron tricarbonyl and (C5H5)TiCl3. Commercially useful examples include (C5H5)Co(CO)2, which is used in the synthesis of substituted pyridines, and methylcyclopentadienyl manganese tricarbonyl, an antiknock agent in petrol.
