
A chemical bond is a lasting attraction between atoms, ions or molecules that enables the formation of molecules. The bond may result from the electrostatic force between oppositely charged ions as in ionic bonds or through the sharing of electrons as in covalent bonds. The strength of chemical bonds varies considerably; there are "strong bonds" or "primary bonds" such as covalent, ionic and metallic bonds, and "weak bonds" or "secondary bonds" such as dipole–dipole interactions, the London dispersion force and hydrogen bonding.
Electronegativity, symbolized as χ, is the tendency for an atom of a given chemical element to attract shared electrons when forming a chemical bond. An atom's electronegativity is affected by both its atomic number and the distance at which its valence electrons reside from the charged nucleus. The higher the associated electronegativity, the more an atom or a substituent group attracts electrons. Electronegativity serves as a simple way to quantitatively estimate the bond energy, and the sign and magnitude of a bond's chemical polarity, which characterizes a bond along the continuous scale from covalent to ionic bonding. The loosely defined term electropositivity is the opposite of electronegativity: it characterizes an element's tendency to donate valence electrons.
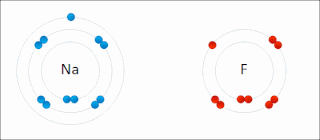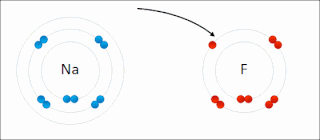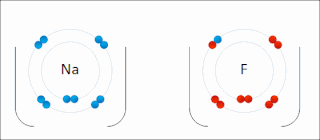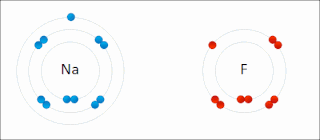
Ionic bonding is a type of chemical bonding that involves the electrostatic attraction between oppositely charged ions, or between two atoms with sharply different electronegativities, and is the primary interaction occurring in ionic compounds. It is one of the main types of bonding along with covalent bonding and metallic bonding. Ions are atoms with an electrostatic charge. Atoms that gain electrons make negatively charged ions. Atoms that lose electrons make positively charged ions. This transfer of electrons is known as electrovalence in contrast to covalence. In the simplest case, the cation is a metal atom and the anion is a nonmetal atom, but these ions can be of a more complex nature, e.g. molecular ions like NH+
4 or SO2−
4. In simpler words, an ionic bond results from the transfer of electrons from a metal to a non-metal in order to obtain a full valence shell for both atoms.
In electrophilic aromatic substitution reactions, existing substituent groups on the aromatic ring influence the overall reaction rate or have a directing effect on positional isomer of the products that are formed. An electron donating group (EDG) or electron releasing group is an atom or functional group that donates some of its electron density into a conjugated π system via resonance (mesomerism) or inductive effects —called +M or +I effects, respectively—thus making the π system more nucleophilic. As a result of these electronic effects, an aromatic ring to which such a group is attached is more likely to participate in electrophilic substitution reaction. EDGs are therefore often known as activating groups, though steric effects can interfere with the reaction.
In chemistry, a hypervalent molecule is a molecule that contains one or more main group elements apparently bearing more than eight electrons in their valence shells. Phosphorus pentachloride, sulfur hexafluoride, chlorine trifluoride, the chlorite ion, and the triiodide ion are examples of hypervalent molecules.

Valence shell electron pair repulsion (VSEPR) theory, is a model used in chemistry to predict the geometry of individual molecules from the number of electron pairs surrounding their central atoms. It is also named the Gillespie-Nyholm theory after its two main developers, Ronald Gillespie and Ronald Nyholm.

Steric effects arise from the spatial arrangement of atoms. When atoms come close together there is a rise in the energy of the molecule. Steric effects are nonbonding interactions that influence the shape (conformation) and reactivity of ions and molecules. Steric effects complement electronic effects, which dictate the shape and reactivity of molecules. Steric repulsive forces between overlapping electron clouds result in structured groupings of molecules stabilized by the way that opposites attract and like charges repel.

In chemistry, conformational isomerism is a form of stereoisomerism in which the isomers can be interconverted just by rotations about formally single bonds. While any two arrangements of atoms in a molecule that differ by rotation about single bonds can be referred to as different conformations, conformations that correspond to local minima on the potential energy surface are specifically called conformational isomers or conformers. Conformations that correspond to local maxima on the energy surface are the transition states between the local-minimum conformational isomers. Rotations about single bonds involve overcoming a rotational energy barrier to interconvert one conformer to another. If the energy barrier is low, there is free rotation and a sample of the compound exists as a rapidly equilibrating mixture of multiple conformers; if the energy barrier is high enough then there is restricted rotation, a molecule may exist for a relatively long time period as a stable rotational isomer or rotamer. When the time scale for interconversion is long enough for isolation of individual rotamers, the isomers are termed atropisomers. The ring-flip of substituted cyclohexanes constitutes another common form of conformational isomerism.

In chemistry, a trigonal bipyramid formation is a molecular geometry with one atom at the center and 5 more atoms at the corners of a triangular bipyramid. This is one geometry for which the bond angles surrounding the central atom are not identical, because there is no geometrical arrangement with five terminal atoms in equivalent positions. Examples of this molecular geometry are phosphorus pentafluoride, and phosphorus pentachloride in the gas phase.

In chemistry, pi stacking refers to the presumptive attractive, noncovalent interactions between the pi bonds of aromatic rings. However this is a misleading description of the phenomena since direct stacking of aromatic rings is electrostatically repulsive. What is more commonly observed is either a staggered stacking or pi-teeing interaction both of which are electrostatic attractive For example, the most commonly observed interactions between aromatic rings of amino acid residues in proteins is a staggered stacked followed by a perpendicular orientation. Sandwiched orientations are relatively rare.
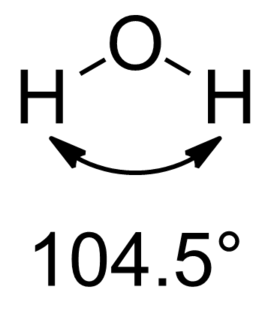
In chemistry, Bent's rule describes and explains the relationship between the orbital hybridization of central atoms in molecules and the electronegativities of substituents. The rule was stated by Henry A. Bent as follows:
Atomic s character concentrates in orbitals directed toward electropositive substituents.

In organic chemistry, the anomeric effect or Edward-Lemieux effect is a stereoelectronic effect that describes the tendency of heteroatomic substituents adjacent to a heteroatom within a cyclohexane ring to prefer the axial orientation instead of the less hindered equatorial orientation that would be expected from steric considerations. This effect was originally observed in pyranose rings by J. T. Edward in 1955 when studying carbohydrate chemistry.

In the study of conformational isomerism, the Gauche effect is an atypical situation where a gauche conformation is more stable than the anti conformation (180°).
Physical organic chemistry, a term coined by Louis Hammett in 1940, refers to a discipline of organic chemistry that focuses on the relationship between chemical structures and reactivity, in particular, applying experimental tools of physical chemistry to the study of organic molecules. Specific focal points of study include the rates of organic reactions, the relative chemical stabilities of the starting materials, reactive intermediates, transition states, and products of chemical reactions, and non-covalent aspects of solvation and molecular interactions that influence chemical reactivity. Such studies provide theoretical and practical frameworks to understand how changes in structure in solution or solid-state contexts impact reaction mechanism and rate for each organic reaction of interest.

In coordination chemistry the bite angle is the ligand–metal–ligand bond angle of coordination complex containing a bidentate ligand. This geometric parameter is used to classify chelating ligands, including those in organometallic complexes. It is most often discussed in terms of catalysis, as changes in bite angle can affect not just the activity and selectivity of a catalytic reaction but even allow alternative reaction pathways to become accessible.
This glossary of chemistry terms is a list of terms and definitions relevant to chemistry, including chemical laws, diagrams and formulae, laboratory tools, glassware, and equipment. Chemistry is a physical science concerned with the composition, structure, and properties of matter, as well as the changes it undergoes during chemical reactions; it features an extensive vocabulary and a significant amount of jargon.
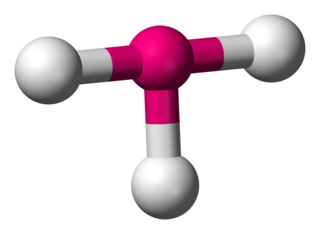
In chemistry, T-shaped molecular geometry describes the structures of some molecules where a central atom has three ligands. Ordinarily, three-coordinated compounds adopt trigonal planar or pyramidal geometries. Examples of T-shaped molecules are the halogen trifluorides, such as ClF3.
A-Values are numerical values used in the determination of the most stable orientation of atoms in a molecule, as well as a general representation of steric bulk. A-values are derived from energy measurements of the different cyclohexane conformations of a monosubstituted cyclohexane chemical. Substituents on a cyclohexane ring prefer to reside in the equatorial position to the axial. The difference in Gibbs free energy (ΔG) between the higher energy conformation and the lower energy conformation is the A-value for that particular substituent.
Iodane generally refers to any organic derivative of iodine. Without modifier, iodane is the systematic name for the parent hydride of iodine, HI. Thus, any organoiodine compound with general formula RI is a substituted iodane. However, as used in the context of organic synthesis, the term iodane more specifically refers to organoiodine compounds with nonstandard bond order of bonds between iodine and other atoms, i.e., bond order of iodine greater than 1, making this term a synonym for hypervalent iodine. These iodine compounds are hypervalent because the iodine atom formally contains more than the 8 electrons in the valence shell required for the octet rule. When iodine is ligated to an organic residue and electronegative ligands, hypervalent iodine occurs in a +3 oxidation state as iodine(III) or λ3-iodane, or in a +5 oxidation state as iodine(V) or λ5-iodane, or in a +7 oxidation state as iodine(VII) or λ7-iodane. Here, lambda convention is used to give the nonstandard bond order.

An oxocarbeniumion is a chemical species characterized by a central sp2-hybridized carbon, an oxygen substituent, and an overall positive charge that is delocalized between the central carbon and oxygen atoms. An oxocarbenium ion is represented by two limiting resonance structures, one in the form of a carbenium ion with the positive charge on carbon and the other in the form of an oxonium species with the formal charge on oxygen. As a resonance hybrid, the true structure falls between the two. Compared to neutral carbonyl compounds like ketones or esters, the carbenium ion form is a larger contributor to the structure. They are common reactive intermediates in the hydrolysis of glycosidic bonds, and are a commonly used strategy for chemical glycosylation. These ions have since been proposed as reactive intermediates in a wide range of chemical transformations, and have been utilized in the total synthesis of several natural products. In addition, they commonly appear in mechanisms of enzyme-catalyzed biosynthesis and hydrolysis of carbohydrates in nature. Anthocyanins are natural flavylium dyes, which are stabilized oxocarbenium compounds. Anthocyanins are responsible for the colors of a wide variety of common flowers such as pansies and edible plants such as eggplant and blueberry.
 PClF4
PClF4 PCl2F3
PCl2F3 PCl3F2
PCl3F2 PCl4F
PCl4F


