Interaction of an organic molecule's reaction center with unconjugated electrons
In organic chemistry, neighbouring group participation (NGP, also known as anchimeric assistance) has been defined by the International Union of Pure and Applied Chemistry (IUPAC) as the interaction of a reaction centre with a lone pair of electrons in an atom or the electrons present in a sigma or pi bond contained within the parent molecule but not conjugated with the reaction centre.[1][2][3][4] When NGP is in operation it is normal for the reaction rate to be increased. It is also possible for the stereochemistry of the reaction to be abnormal (or unexpected) when compared with a normal reaction. While it is possible for neighbouring groups to influence many reactions in organic chemistry (e.g. the reaction of a diene such as 1,3-cyclohexadiene with maleic anhydride normally gives the endo isomer because of a secondary effect {overlap of the carbonyl group π orbitals with the transition state in the Diels-Alder reaction}) this page is limited to neighbouring group effects seen with carbocations and SN2 reactions.
In this type of substitution reaction, one group of the substrate participates initially in the reaction and thereby affects the reaction. A classic example of NGP is the reaction of a sulfur or nitrogen mustard with a nucleophile, the rate of reaction is much higher for the sulfur mustard and a nucleophile than it would be for a primary or secondary alkylchloride without a heteroatom.[5]
Ph−S−CH2−CH2−Cl reacts with water 600 times faster than CH3−CH2−CH2−Cl.[5]
NGP by an alkene
The π orbitals of an alkene can stabilize a transition state by helping to delocalize the positive charge of the carbocation. For instance the unsaturatedtosylate will react more quickly (1011 times faster for aqueous solvolysis) with a nucleophile than the saturated tosylate.
The carbocationic intermediate will be stabilized by resonance where the positive charge is spread over several atoms. In the diagram below this is shown.
Here is a different view of the same intermediates.
Even if the alkene is more remote from the reacting center the alkene can still act in this way. For instance in the following alkyl benzenesulfonate the alkene is able to delocalise the carbocation.
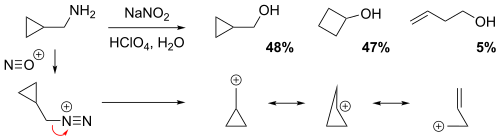
NGP by a cyclopropane, cyclobutane or a homoallyl group
The reaction of cyclopropylmethamine with sodium nitrite in dilute aqueous perchloric acid solution yielded a mixture of 48% cyclopropylmethyl alcohol, 47% cyclobutanol, and 5% homoallylic alcohol (but-3-en-1-ol).[6] In the non-classical perspective, the positive charge is delocalized throughout the carbocation intermediate structure via resonance, resulting in partial (electron-deficient) bonds. Evidently, the relatively low yield of the homoallylic alcohol implies that the homoallylic structure is the weakest resonance contributor.
NGP by an aromatic ring
An aromatic ring can assist in the formation of a carbocationic intermediate called a phenonium ion by delocalising the positive charge.
When the following tosylate reacts with acetic acid in solvolysis then rather than a simple SN2 reaction forming B, a 48:48:4 mixture of A, B (which are enantiomers) and C+D was obtained.[7][8]
The mechanism which forms A and B is shown below.
NGP by aliphatic C-C or C-H bonds
Aliphatic C-C or C-H bonds can lead to charge delocalization if these bonds are close and antiperiplanar to the leaving group. Corresponding intermediates are referred to a nonclassical ions, with the 2-norbornyl system as the most well known case.
↑ Stalford, Susanne A.; Kilner, Colin A.; Leach, Andrew G.; Turnbull, W. Bruce (2009-12-07). "Neighbouring group participation vs. addition to oxacarbenium ions: studies on the synthesis of mycobacterial oligosaccharides". Organic & Biomolecular Chemistry. 7 (23). Royal Society of Chemistry: 4842–4852. doi:10.1039/B914417J. PMID19907773.
↑ Bowden, Keith (1993). "Neighbouring Group Participation by Carbonyl Groups in Ester Hydrolysis". Advances in Physical Organic Chemistry. Elsevier. doi:10.1016/S0065-3160(08)60182-3.
1 2 Clayden, Jonathan; Greeves, Nick; Warren, Stuart G. (2012). Organic chemistry (2nded.). Oxford; New YorK: Oxford University Press. p.932. ISBN978-0-19-927029-3.
↑ Cram, Donald J. (December 1949). "Studies in Stereochemistry. I. The Stereospecific Wagner--Meerwein Rearrangement of the Isomers of 3-Phenyl-2-butanol". Journal of the American Chemical Society. 71 (12): 3863–3870. Bibcode:1949JAChS..71.3863C. doi:10.1021/ja01180a001.
↑ Cram, Donald J. (May 1952). "Studies in Stereochemistry. V. Phenonium Sulfonate Ion-pairs as Intermediates in the Intramolecular Rearrangements and Solvolysis Reactions that Occur in the 3-Phenyl-2-butanol System". Journal of the American Chemical Society. 74 (9): 2129–2137. doi:10.1021/ja01129a001.
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.