Sharpless asymmetric dihydroxylation is the chemical reaction of an alkene with osmium tetroxide in the presence of a chiral quinine ligand to form a vicinal diol. The reaction has been applied to alkenes of virtually every substitution, often high enantioselectivities are realized, with the chiral outcome controlled by the choice of dihydroquinidine (DHQD) vs dihydroquinine (DHQ) as the ligand. Asymmetric dihydroxylation reactions are also highly site selective, providing products derived from reaction of the most electron-rich double bond in the substrate.
The Heck reaction is the chemical reaction of an unsaturated halide with an alkene in the presence of a base and a palladium catalyst to form a substituted alkene. It is named after Tsutomu Mizoroki and Richard F. Heck. Heck was awarded the 2010 Nobel Prize in Chemistry, which he shared with Ei-ichi Negishi and Akira Suzuki, for the discovery and development of this reaction. This reaction was the first example of a carbon-carbon bond-forming reaction that followed a Pd(0)/Pd(II) catalytic cycle, the same catalytic cycle that is seen in other Pd(0)-catalyzed cross-coupling reactions. The Heck reaction is a way to substitute alkenes.

In organic chemistry, the ene reaction is a chemical reaction between an alkene with an allylic hydrogen and a compound containing a multiple bond, in order to form a new σ-bond with migration of the ene double bond and 1,5 hydrogen shift. The product is a substituted alkene with the double bond shifted to the allylic position.
The 1,3-dipolar cycloaddition is a chemical reaction between a 1,3-dipole and a dipolarophile to form a five-membered ring. The earliest 1,3-dipolar cycloadditions were described in the late 19th century to the early 20th century, following the discovery of 1,3-dipoles. Mechanistic investigation and synthetic application were established in the 1960s, primarily through the work of Rolf Huisgen. Hence, the reaction is sometimes referred to as the Huisgen cycloaddition. 1,3-dipolar cycloaddition is an important route to the regio- and stereoselective synthesis of five-membered heterocycles and their ring-opened acyclic derivatives. The dipolarophile is typically an alkene or alkyne, but can be other pi systems. When the dipolarophile is an alkyne, aromatic rings are generally produced.
In chemistry, stereoselectivity is the property of a chemical reaction in which a single reactant forms an unequal mixture of stereoisomers during a non-stereospecific creation of a new stereocenter or during a non-stereospecific transformation of a pre-existing one. The selectivity arises from differences in steric and electronic effects in the mechanistic pathways leading to the different products. Stereoselectivity can vary in degree but it can never be total since the activation energy difference between the two pathways is finite: both products are at least possible and merely differ in amount. However, in favorable cases, the minor stereoisomer may not be detectable by the analytic methods used.
Organopalladium chemistry is a branch of organometallic chemistry that deals with organic palladium compounds and their reactions. Palladium is often used as a catalyst in the reduction of alkenes and alkynes with hydrogen. This process involves the formation of a palladium-carbon covalent bond. Palladium is also prominent in carbon-carbon coupling reactions, as demonstrated in tandem reactions.
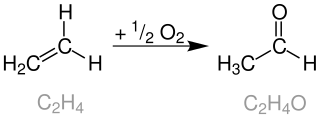
The Wacker process or the Hoechst-Wacker process refers to the oxidation of ethylene to acetaldehyde in the presence of palladium(II) chloride and copper(II) chloride as the catalyst. This chemical reaction was one of the first homogeneous catalysis with organopalladium chemistry applied on an industrial scale.
Dihydroxylation is the process by which an alkene is converted into a vicinal diol. Although there are many routes to accomplish this oxidation, the most common and direct processes use a high-oxidation-state transition metal. The metal is often used as a catalyst, with some other stoichiometric oxidant present. In addition, other transition metals and non-transition metal methods have been developed and used to catalyze the reaction.
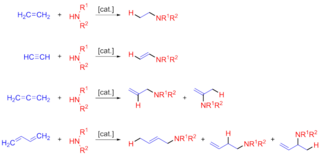
In organic chemistry, hydroamination is the addition of an N−H bond of an amine across a carbon-carbon multiple bond of an alkene, alkyne, diene, or allene. In the ideal case, hydroamination is atom economical and green. Amines are common in fine-chemical, pharmaceutical, and agricultural industries. Hydroamination can be used intramolecularly to create heterocycles or intermolecularly with a separate amine and unsaturated compound. The development of catalysts for hydroamination remains an active area, especially for alkenes. Although practical hydroamination reactions can be effected for dienes and electrophilic alkenes, the term hydroamination often implies reactions metal-catalyzed processes.

Strychnine total synthesis in chemistry describes the total synthesis of the complex biomolecule strychnine. The first reported method by the group of Robert Burns Woodward in 1954 is considered a classic in this research field.
A (4+3) cycloaddition is a cycloaddition between a four-atom π-system and a three-atom π-system to form a seven-membered ring. Allyl or oxyallyl cations (propenylium-2-olate) are commonly used three-atom π-systems, while a diene plays the role of the four-atom π-system. It represents one of the relatively few synthetic methods available to form seven-membered rings stereoselectively in high yield.
Ketene cycloadditions are the reactions of the pi system of ketenes with unsaturated compounds to provide four-membered or larger rings. [2+2], [3+2], and [4+2] variants of the reaction are known.
The nitrone-olefin (3+2) cycloaddition reaction is the combination of a nitrone with an alkene or alkyne to generate an isoxazoline or isoxazolidine via a (3+2) cycloaddition process. This reaction is a 1,3-dipolar cycloaddition, in which the nitrone acts as the 1,3-dipole, and the alkene or alkyne as the dipolarophile.
The intramolecular Heck reaction (IMHR) in chemistry is the coupling of an aryl or alkenyl halide with an alkene in the same molecule. The reaction may be used to produce carbocyclic or heterocyclic organic compounds with a variety of ring sizes. Chiral palladium complexes can be used to synthesize chiral intramolecular Heck reaction products in non-racemic form.
In organic chemistry, the Baylis–Hillman, Morita–Baylis–Hillman, or MBH reaction is a carbon-carbon bond-forming reaction between an activated alkene and a carbon electrophile in the presence of a nucleophilic catalyst, such as a tertiary amine or phosphine. The product is densely functionalized, joining the alkene at the α-position to a reduced form of the electrophile.
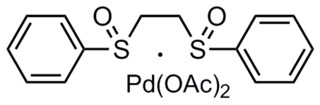
The White catalyst is a transition metal coordination complex named after the chemist by whom it was first synthesized, M. Christina White, a professor at the University of Illinois. The catalyst has been used in a variety of allylic C-H functionalization reactions of α-olefins. In addition, it has been shown to catalyze oxidative Heck reactions.
The Tsuji–Trost reaction is a palladium-catalysed substitution reaction involving a substrate that contains a leaving group in an allylic position. The palladium catalyst first coordinates with the allyl group and then undergoes oxidative addition, forming the π-allyl complex. This allyl complex can then be attacked by a nucleophile, resulting in the substituted product.
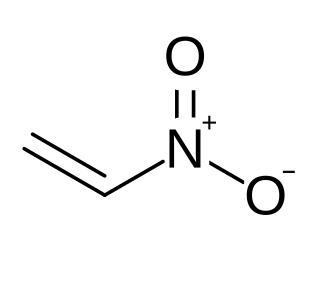
Nitroethylene (also known as nitroethene) is a liquid organic compound with the formula C2H3NO2. It is the simplest nitroalkene, which are unsaturated carbon chains with at least one double bond and a NO2 functional group. Nitroethylene serves as a useful intermediate in the production of various other chemicals.

Dynamic kinetic resolution in chemistry is a type of kinetic resolution where 100% of a racemic compound can be converted into an enantiopure compound. It is applied in asymmetric synthesis. Asymmetric synthesis has become a much explored field due to the challenge of creating a compound with a single 3D structure. Even more challenging is the ability to take a racemic mixture and have only one chiral product left after a reaction. One method that has become an exceedingly useful tool is dynamic kinetic resolution (DKR). DKR utilizes a center of a particular molecule that can be easily epimerized so that the (R) and (S) enantiomers can interconvert throughout the reaction process. At this point the catalyst can selectively lower the transition state energy of a single enantiomer, leading to almost 100% yield of one reaction pathway over the other. The figure below is an example of an energy diagram for a compound with an (R) and (S) isomer.
Heterobimetallic catalysis is an approach to catalysis that employs two different metals to promote a chemical reaction. Included in this definition are cases where: 1) each metal activates a different substrate, 2) both metals interact with the same substrate, and 3) only one metal directly interacts with the substrate(s), while the second metal interacts with the first.














