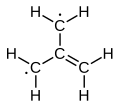
Trimethylenemethane, average of three configurations. Formally, the radial bonds have valency 4/3. Each terminal carbon has 2/3 of an unfilled valence bond.
The electronic structure of trimethylenemethane was discussed in 1948.[1][2] It is a neutral four-carbon molecule containing four pi molecular orbitals. When trapped in a solid matrix at about 90K (−183°C), the six hydrogen atoms of the molecule are equivalent. Thus, it can be described either as zwitterion, or as the simplest conjugated hydrocarbon that cannot be given a Kekulé structure. It can be described as the superposition of three states:
It has a tripletground state (3A2′/3B2), and is therefore a diradical in the stricter sense of the term.[3] Calculations predict a planar molecule with three-fold rotational symmetry, with approximate bond lengths 1.40Å (C–C) and 1.08Å (C–H). The H–C–H angle in each methylene is about 121°.[1]
Of the three singlet excited states, the first one, 11A1 (1.17eV above ground), is a closed shell diradical with flat geometry and fully degenerate threefold (D3h) symmetry. The second one, 11B2 (also at 1.17eV), is an open-shell radical with a D3h-symmetric equilibrium between three equal geometries; each has a longer C–C bond (1.48Å) and two shorter ones (1.38Å), and is flat and bilaterally symmetric except that the longer methylene is twisted 79° out of the plane (C2 symmetry). The third singlet state, 21A1/1A1′ (3.88eV), is also a D3h-symmetric equilibrium of three geometries; each is planar with one shorter C–C bond and two longer ones (C2ν symmetry).[1]
The next higher energy states are degenerate triplets, 13A1 and 23B2 (4.61eV), with one excited electron; and a quintet state, 5B2 (7.17eV), with the p orbitals occupied by single electrons and D3h symmetry.
Preparation
Trimethylenemethane was first obtained from photolysis of the diazo compound 4-methylene-Δ1-pyrazoline with expulsion of nitrogen, in a frozen dilute glassy solution at −196°C (77K).[3]
Trimethylenemethane has been obtained also by treating potassium with 2-iodomethyl-3-iodopropene and isobutylene diiodide (IH 2C)2C=CH 2 in the gas phase. However the product quickly dimerizes to yield 1,4-dimethylenecyclohexane, and also 2-methylpropene by abstracting two hydrogen atoms from other molecules (hydrocarbon or potassium hydride).[4]
Three classes of compounds have been used to generate synthetically useful TMM-derivative reaction intermediates: diazenes, silyl-substituted allylicacetates and methylenecyclopropenes. In the first case, bridged diazenes are used to avoid competitive closure to MCPs and dimerization reactions.[5] The latter case requires stabilization of a zwitterion, as with e.g. acetal 1:[6]
Alternatively, palladium(0) or nickel(0) catalysts can stabilize the zwitterion:[7]
Silylated allylic acetates, carbonates and other substituted allyl compounds may form TMM synthons under palladium catalysis.[8]
TMM complexes have been examined for their potential in organic synthesis, specifically in the trimethylenemethane cycloaddition reaction (see §Cycloaddition) with only modest success. One example is a palladium-catalyzed [3+2]cycloaddition of trimethylenemethane.[10][5]
Structure of Ru(trimethylenemethane)(CO)3, viewed down C3 axis.[6]
Structure of Ru(trimethylenemethane)(CO)3, viewed orthogonal to C3 axis.
Organic reactions
Unligated trimethylenemethanes are unstable, and rapidly close a ring to methylidenecyclopropanes.[5] The problem is generally less severe for five-membered, cyclic TMMs due to ring strain in the corresponding methylidenecyclopropanes.
Cycloaddition
Trimethylenemethane cycloaddition is the formal (3+2) annulation of trimethylenemethane (TMM) derivatives to two-atom pi systems. Although TMM itself is too reactive and unstable to be stored, reagents which can generate TMM or TMM synthonsin situ can be used to effect cycloaddition reactions with appropriate electron acceptors. Generally, electron-deficient pi bonds undergo cyclization with TMMs more easily than electron-rich pi bonds.[6]
Usually, unless a cyclic pi system is involved TMM cycloadditions exhibit 2π periselectivity and do not react with larger pi systems. Polar MCPs, for example, react only with the 2,3 double bond of polyunsaturated esters.[6]
TMM's singlet and triplet states exhibit different reactivity and selectivity profiles.[5] A singlet (3+2) cycloaddition, when it is concerted, is believed to proceed under frontier orbital control. When electron-rich TMMs are involved, the A orbital serves as the HOMO (leading to fused products if the TMM is cyclic). When electron-poor (or unsubstituted) TMMs are involved, the S orbital serves as the HOMO (leading to bridged products if the TMM is cyclic). Cycloadditions involving the triplet state are stepwise, and usually result in configurational scrambling in the two-atom component.[6]
Diazene-derived TMMs cyclize with an alkenic acceptor to either fused or bridged products.[6] Fused products are generally favored, unless the methylene carbon bears electron-donating groups. The configuration of the alkene is maintained as long as the reaction is proceeding through a singlet TMM.[8]
Unless catalyzed by transition metals, methylidenecyclopropane opening is also stereospecific with respect to alkene geometry, and exhibits high selectivity for endo products in most cases.
With catalysis, cyclization takes place in a stepwise fashion and does not exhibit stereospecificity. Rapid racemization of chiral π-allyl palladium complexes occurs, and only moderate diastereoselectivity is observed in reactions of chiral allylic acetates. Chiral non-racemic alkenes, however, may exhibit moderate to high diastereoselectivity. The reaction is highly regioselective, providing only the substitution pattern shown below regardless of the position of the R' group on the starting allylic acetate.
Chiral auxiliaries on the alkene partner have been used for stereoselective transformations. In the reaction of camphorsultam-derived unsaturated amides, lower temperatures were needed to achieve high selectivities.[11]
In reactions of silyl-substituted allylic acetates, chiral sulfoxides can be used to enforce high diastereofacial selectivity.[12]
Carbonyl compounds may be used as the 2π component under the appropriate conditions. For instance, in the presence of an indium co-catalyst, the reactive 2π component of the cycloaddition below switches from the C-C to the C-O double bond.[13]
Polarized trimethylenemethanes generated from polar MCPs are also useful substrates for (3+2) reactions with polar double bonds as the 2π component. Orthoester products are generally favored over ketene acetals.[14]
Comparison with other methods
Although 1,3-dipolar cycloaddition is a useful method for the generation of five-membered heterocyclic compounds, few methods exist to synthesize five-membered carbocyclic rings in a single step via annulation. Most of these, like TMM cycloaddition, rely on the generation of a suitable three-atom component for combination with a stable two-atom partner such as an alkene or alkyne. When heated, cyclopropene acetals rearrange to vinylcarbenes, which can serve as the three-atom component in cycloadditions with highly electron-deficient alkenes.[15]Zinc homoenolates can also serve as indirect three-atom components, and undergo cyclization to cyclopentenones in the presence of an unsaturated ester.[16] Tandem intermolecular-intramolecular cyclization of homopropargylic radicals leads to methylenecyclopropanes.[17]
References
123Slipchenko Lyudmila V., Krylov Anna I. (2003). "Electronic structure of the trimethylenemethane diradical in its ground and electronically excited states: Bonding, equilibrium geometries, and Vibrational frequencies". Journal of Chemical Physics. 118 (15): 6874–6883. Bibcode:2003JChPh.118.6874S. doi:10.1063/1.1561052. S2CID4204676.
↑C. A. Coulson (1948), Journal de Chimie Physique et de Physico-Chimie Biologique, volume 45, page 243. Cited by Slipchenko and Krylov (2003)
1234Paul Dowd (1972). "Trimethylenemethane". Accounts of Chemical Research. 5 (7): 242–248. doi:10.1021/ar50055a003.
123456Nakamura, E.; Yamago, S.; Ejiri, S.; Dorigo, A. E.; Morokuma, K. J. Am. Chem. Soc.1991, 113, 3183.
↑Binger, P.; Büch, H. M. Top. Curr. Chem.1987, 135, 77.
12Trost, B. M. Angew. Chem. Int. Ed. Engl.1986, 25, 1.
12J. S. Ward & R. Pettit (1970). "Trimethylenemethane complexes of iron, molybdenum, and chromium". Journal of the Chemical Society D (21): 1419–1420. doi:10.1039/C29700001419.
↑Barry M. Trost (1979). "New conjunctive reagents. 2-Acetoxymethyl-3-allyltrimethylsilane for methylenecyclopentane annulations catalyzed by palladium(0)". Journal of the American Chemical Society. 101 (21): 6429–6432. Bibcode:1979JAChS.101.6429T. doi:10.1021/ja00515a046.
↑Binger, P.; Schäfer, B. Tetrahedron Lett.1988, 29, 529.
↑Chaigne, F.; Gotteland, J.-P.; Malacria, M. Tetrahedron Lett.1989, 30, 1803.
↑Trost, B. M.; Sharma, S.; Schmidt, T. J. Am. Chem. Soc.1992, 114, 7903.
↑Yamago, S.; Nakamura, E. J. Org. Chem.1990, 55, 5553.
↑Boger, D. L.; Brotherton, C. E. J. Am. Chem. Soc.1986, 108, 6695.
↑Crimmins, M. T.; Nantermet, P. G. J. Org. Chem.1990, 55, 4235.
↑Curran, D. P.; Chen, M.-H. J. Am. Chem. Soc.1987, 109, 6558.
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.