The reaction is notable for being among the first reported catalytic cross-coupling methods. Despite the subsequent development of alternative reactions (Suzuki, Sonogashira, Stille, Hiyama, Negishi), the Kumada coupling continues to be employed in many synthetic applications, including the industrial-scale production of aliskiren, a hypertension medication, and polythiophenes, useful in organic electronic devices.
History
The first investigations into the catalytic coupling of Grignard reagents with organic halides date back to the 1941 study of cobalt catalysts by Morris S. Kharasch and E. K. Fields.[3] In 1971, Tamura and Kochi elaborated on this work in a series of publications demonstrating the viability of catalysts based on silver,[4] copper[5] and iron.[6] However, these early approaches produced poor yields due to substantial formation of homocoupling products, where two identical species are coupled.
These efforts culminated in 1972, when the Corriu and Kumada groups concurrently reported the use of nickel-containing catalysts. With the introduction of palladium catalysts in 1975 by the Murahashi group, the scope of the reaction was further broadened.[7] Subsequently, many additional coupling techniques have been developed, culminating in the 2010 Nobel Prize in Chemistry recognized Ei-ichi Negishi, Akira Suzuki and Richard F. Heck for their contributions to the field.
Mechanism
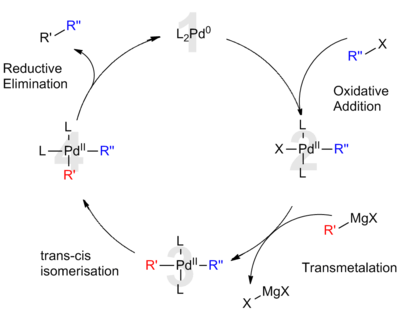
Accepted catalytic cycle for Kumada cross coupling reaction
Palladium catalysis
According to the widely accepted mechanism, the palladium-catalyzed Kumada coupling is understood to be analogous to palladium's role in other cross coupling reactions. The proposed catalytic cycle involves both palladium(0) and palladium(II) oxidation states. Initially, the electron-rich Pd(0) catalyst (1) inserts into the R–X bond of the organic halide. This oxidative addition forms an organo-Pd(II)-complex (2). Subsequent transmetalation with the Grignard reagent forms a hetero-organometallic complex (3). Before the next step, isomerization is necessary to bring the organic ligands next to each other into mutually cis positions. Finally, reductive elimination of (4) forms a carbon–carbon bond and releases the cross coupled product while regenerating the Pd(0) catalyst (1).[8] For palladium catalysts, the frequently rate-determining oxidative addition occurs more slowly than with nickel catalyst systems.[8]
Nickel catalysis
Example of Kumada's cross coupling with Nickel
Current understanding of the mechanism for the nickel-catalyzed coupling is limited. Indeed, the reaction mechanism is believed to proceed differently under different reaction conditions and when using different nickel ligands.[9] In general the mechanism can still be described as analogous to the palladium scheme (right). Under certain reaction conditions, however, the mechanism fails to explain all observations. Examination by Vicic and coworkers using tridentate terpyridine ligand identified intermediates of a Ni(II)-Ni(I)-Ni(III) catalytic cycle,[10] suggesting a more complicated scheme. Additionally, with the addition of butadiene, the reaction is believed to involve a Ni(IV) intermediate.[11]
Scope
Organic halides and pseudohalides
The Kumada coupling has been successfully demonstrated for a variety of aryl or vinyl halides. In place of the halide reagent pseudohalides can also be used, and the coupling has been shown to be quite effective using tosylate[12] and triflate[13] species in variety of conditions.
Despite broad success with aryl and vinyl couplings, the use of alkyl halides is less general due to several complicating factors. Having no π-electrons, alkyl halides require different oxidative addition mechanisms than aryl or vinyl groups, and these processes are currently poorly understood.[9] Additionally, the presence of β-hydrogens makes alkyl halides susceptible to competitive elimination processes.[14]
These issues have been circumvented by the presence of an activating group, such as the carbonyl in α-bromoketones, that drives the reaction forward. However, Kumada couplings have also been performed with non-activated alkyl chains, often through the use of additional catalysts or reagents. For instance, with the addition of 1,3-butadienes Kambe and coworkers demonstrated nickel catalyzed alkyl–alkyl couplings that would otherwise be unreactive.[15]
Though poorly understood, the mechanism of this reaction is proposed to involve the formation of an octadienyl nickel complex. This catalyst is proposed to undergo transmetalation with a Grignard reagent first, prior to the reductive elimination of the halide, reducing the risk of β-hydride elimination. However, the presence of a Ni(IV) intermediate is contrary to mechanisms proposed for aryl or vinyl halide couplings.[11]
Proposed Kumada coupling mechanism with addition of butadiene
Grignard reagent
Couplings involving aryl and vinyl Grignard reagents were reported in the original publications by Kumada and Corriu.[2] Alkyl Grignard reagents can also be used without difficulty, as they do not suffer from β-hydride elimination processes. Although the Grignard reagent inherently has poor functional group tolerance, low-temperature syntheses have been prepared with highly functionalized aryl groups.[16]
Catalysts
Kumada couplings can be performed with a variety of nickel(II) or palladium(II) catalysts. The structures of the catalytic precursors can be generally formulated as ML2X2, where L is a phosphine ligand.[17] Common choices for L2 include bidentate diphosphine ligands such as dppe and dppp among others.
Work by Alois Fürstner and coworkers on iron-based catalysts have shown reasonable yields. The catalytic species in these reactions is proposed to be an "inorganic Grignard reagent" consisting of Fe(MgX)2.[18]
Reaction conditions
The reaction typically is carried out in tetrahydrofuran or diethyl ether as solvent. Such ethereal solvents are convenient because these are typical solvents for generating the Grignard reagent.[2] Due to the high reactivity of the Grignard reagent, Kumada couplings have limited functional group tolerance which can be problematic in large syntheses. In particular, Grignard reagents are sensitive to protonolysis from even mildly acidic groups such as alcohols. They also add to carbonyls and other oxidative groups.
As in many coupling reactions, the transition metal palladium catalyst is often air-sensitive, requiring an inert Argon or nitrogen reaction environment.
Both cis- and trans-olefin halides promote the overall retention of geometric configuration when coupled with alkyl Grignards. This observation is independent of other factors, including the choice of catalyst ligands and vinylic substituents.[17]
Conversely, a Kumada coupling using vinylic Grignard reagents proceeds without stereospecificity to form a mixture of cis- and trans-alkenes. The degree of isomerization is dependent on a variety of factors including reagent ratios and the identity of the halide group. According to Kumada, this loss of stereochemistry is attributable to side-reactions between two equivalents of the allylic Grignard reagent.[17]
Asymmetric Kumada couplings can be effected through the use of chiral ligands. Using planar chiralferrocene ligands, enantiomeric excesses (ee) upward of 95% have been observed in aryl couplings.[19] More recently, Gregory Fu and co-workers have demonstrated enantioconvergent couplings of α-bromoketones using catalysts based on bis-oxazoline ligands, wherein the chiral catalyst converts a racemic mixture of starting material to one enantiomer of product with up to 95% ee.[20] The latter reaction is also significant for involving a traditionally inaccessible alkyl halide coupling.
Enantioconvergent coupling of α-bromoketones
Chemoselectivity
Grignard reagents do not typically couple with chlorinated arenes. This low reactivity is the basis for chemoselectivity for nickel insertion into the C–Br bond of bromochlorobenzene using a NiCl2-based catalyst.[21]
NiCl2 catalyzed Kumada coupling shows haloselectivity on bromochlorobenzene.
Applications
Synthesis of aliskiren
The Kumada coupling is suitable for large-scale, industrial processes, such as drug synthesis. The reaction is used to construct the carbon skeleton of aliskiren (trade name Tekturna), a treatment for hypertension.[22]
Kumada coupling in the synthesis of aliskiren
Synthesis of polythiophenes
The Kumada coupling also shows promise in the synthesis of conjugated polymers, polymers such as polyalkylthiophenes (PAT), which have a variety of potential applications in organic solar cells and light-emitting diodes.[23] In 1992, McCollough and Lowe developed the first synthesis of regioregular polyalkylthiophenes by utilizing the Kumada coupling scheme pictured below, which requires subzero temperatures.[24]
Synthesis of polythiophenes via Kumada coupling
Since this initial preparation, the synthesis has been improved to obtain higher yields and operate at room temperature.[25]
↑ Corriu, R. J. P.; Masse, J. P. (1 January 1972). "Activation of Grignard reagents by transition-metal complexes. A new and simple synthesis of trans-stilbenes and polyphenyls". Journal of the Chemical Society, Chemical Communications (3): 144a. doi:10.1039/C3972000144A.
1 2 3 Tamao, Kohei; Sumitani, Koji; Kumada, Makoto (1 June 1972). "Selective carbon–carbon bond formation by cross-coupling of Grignard reagents with organic halides. Catalysis by nickel-phosphine complexes". Journal of the American Chemical Society. 94 (12): 4374–4376. doi:10.1021/ja00767a075.
↑ Kharasch, M. S.; Fields, E. K. (1 September 1941). "Factors Determining the Course and Mechanisms of Grignard Reactions. IV. The Effect of Metallic Halides on the Reaction of Aryl Grignard Reagents and Organic Halides1". Journal of the American Chemical Society. 63 (9): 2316–2320. doi:10.1021/ja01854a006.
↑ Jay K. Kochi and Masuhiko Tamura (1971). "Mechanism of the silver-catalyzed reaction of Grignard reagents with alkyl halides". J. Am. Chem. Soc.93 (6): 1483–1485. doi:10.1021/ja00735a028.
↑ Kochi, Jay K.; Tamura, Masuhiko (1 March 1971). "Alkylcopper(I) in the coupling of Grignard reagents with alkyl halides". Journal of the American Chemical Society. 93 (6): 1485–1487. doi:10.1021/ja00735a029.
↑ Tamura, Masuhiko; Kochi, Jay K. (1 March 1971). "Vinylation of Grignard reagents. Catalysis by iron". Journal of the American Chemical Society. 93 (6): 1487–1489. doi:10.1021/ja00735a030.
↑ Yamamura, Masaaki; Moritani, Ichiro; Murahashi, Shun-Ichi (27 May 1975). "The reaction of σ-vinylpalladium complexes with alkyllithiums. Stereospecific syntheses of olefins from vinyl halides and alkyllithiums". Journal of Organometallic Chemistry. 91 (2): C39–C42. doi:10.1016/S0022-328X(00)89636-9.
1 2 Knappke, Christiane E. I.; Jacobi von Wangelin, Axel (2011). "35 years of palladium-catalyzed cross-coupling with Grignard reagents: how far have we come?". Chemical Society Reviews. 40 (10): 4948–62. doi:10.1039/c1cs15137a. PMID21811712.
↑ Jones, Gavin D.; McFarland, Chris; Anderson, Thomas J.; Vicic, David A. (1 January 2005). "Analysis of key steps in the catalytic cross-coupling of alkyl electrophiles under Negishi-like conditions". Chemical Communications (33): 4211–3. doi:10.1039/b504996b. PMID16100606.
1 2 Frisch, Anja C.; Beller, Matthias (21 January 2005). "Catalysts for Cross-Coupling Reactions with Non-activated Alkyl Halides". Angewandte Chemie International Edition. 44 (5): 674–688. doi:10.1002/anie.200461432. PMID15657966.
↑ Limmert, Michael E.; Roy, Amy H.; Hartwig, John F. (1 November 2005). "Kumada Coupling of Aryl and Vinyl Tosylates under Mild Conditions". The Journal of Organic Chemistry. 70 (23): 9364–9370. doi:10.1021/jo051394l. PMID16268609.
↑ Busacca, Carl A.; Eriksson, Magnus C.; Fiaschi, Rita (1999). "Cross coupling of vinyl triflates and alkyl Grignard reagents catalyzed by nickel(0)-complexes". Tetrahedron Letters. 40 (16): 3101–3104. doi:10.1016/S0040-4039(99)00439-6.
↑ Rudolph, Alena; Lautens, Mark (30 March 2009). "Secondary Alkyl Halides in Transition-Metal-Catalyzed Cross-Coupling Reactions". Angewandte Chemie International Edition. 48 (15): 2656–2670. doi:10.1002/anie.200803611. PMID19173365.
↑ Terao, Jun; Watanabe, Hideyuki; Ikumi, Aki; Kuniyasu, Hitoshi; Kambe, Nobuaki (1 April 2002). "Nickel-Catalyzed Cross-Coupling Reaction of Grignard Reagents with Alkyl Halides and Tosylates: Remarkable Effect of 1,3-Butadienes". Journal of the American Chemical Society. 124 (16): 4222–4223. doi:10.1021/ja025828v. PMID11960446.
↑ Adrio, Javier; Carretero, Juan C. (15 November 2010). "Functionalized Grignard Reagents in Kumada Cross-Coupling Reactions". ChemCatChem. 2 (11): 1384–1386. doi:10.1002/cctc.201000237. S2CID98429919.
↑ Ikoma, Yoshiharu; Ando, Kazuhiko; Naoi, Yoshitake; Akiyama, Takeo; Sugimori, Akira (1 February 1991). "Halogen Selectivity in Nickel Salt-Catalyzed Cross-Coupling of Aryl Grignard Reagents with Bromochlorobenzenes a Novel Synthetic Method of Unsymmetrical Terphenyl". Synthetic Communications. 21 (3): 481–487. doi:10.1080/00397919108016772.
↑ Johnson and Lee (2010). Modern Drug Synthesis. Hoboken, NJ: John Wiley & Sons, Inc. pp.153–154. ISBN978-0-470-52583-8.
↑ Cheng, Yen-Ju; Yang, Sheng-Hsiung; Hsu, Chain-Shu (11 November 2009). "Synthesis of Conjugated Polymers for Organic Solar Cell Applications". Chemical Reviews. 109 (11): 5868–5923. doi:10.1021/cr900182s. PMID19785455.
↑ McCullough, Richard D.; Lowe, Renae D. (1 January 1992). "Enhanced electrical conductivity in regioselectively synthesized poly(3-alkylthiophenes)". Journal of the Chemical Society, Chemical Communications (1): 70. doi:10.1039/C39920000070.
↑ Loewe, Robert S.; Ewbank, Paul C.; Liu, Jinsong; Zhai, Lei; McCullough, Richard D. (1 June 2001). "Regioregular, Head-to-Tail Coupled Poly(3-alkylthiophenes) Made Easy by the GRIM Method: Investigation of the Reaction and the Origin of Regioselectivity". Macromolecules. 34 (13): 4324–4333. Bibcode:2001MaMol..34.4324L. doi:10.1021/ma001677+.
Related Research Articles
The Heck reaction is the chemical reaction of an unsaturated halide with an alkene in the presence of a base and a palladium catalyst to form a substituted alkene. It is named after Tsutomu Mizoroki and Richard F. Heck. Heck was awarded the 2010 Nobel Prize in Chemistry, which he shared with Ei-ichi Negishi and Akira Suzuki, for the discovery and development of this reaction. This reaction was the first example of a carbon-carbon bond-forming reaction that followed a Pd(0)/Pd(II) catalytic cycle, the same catalytic cycle that is seen in other Pd(0)-catalyzed cross-coupling reactions. The Heck reaction is a way to substitute alkenes.
The Stille reaction is a chemical reaction widely used in organic synthesis. The reaction involves the coupling of two organic groups, one of which is carried as an organotin compound. A variety of organic electrophiles provide the other coupling partner. The Stille reaction is one of many palladium-catalyzed coupling reactions.
The Suzuki reaction is an organic reaction, classified as a cross-coupling reaction, where the coupling partners are a boronic acid and an organohalide and the catalyst is a palladium(0) complex. It was first published in 1979 by Akira Suzuki, and he shared the 2010 Nobel Prize in Chemistry with Richard F. Heck and Ei-ichi Negishi for their contribution to the discovery and development of palladium-catalyzed cross-couplings in organic synthesis. This reaction is also known as the Suzuki–Miyaura reaction or simply as the Suzuki coupling. It is widely used to synthesize polyolefins, styrenes, and substituted biphenyls. Several reviews have been published describing advancements and the development of the Suzuki reaction. The general scheme for the Suzuki reaction is shown below, where a carbon-carbon single bond is formed by coupling a halide (R1-X) with an organoboron species (R2-BY2) using a palladium catalyst and a base.
The Sonogashira reaction is a cross-coupling reaction used in organic synthesis to form carbon–carbon bonds. It employs a palladium catalyst as well as copper co-catalyst to form a carbon–carbon bond between a terminal alkyne and an aryl or vinyl halide.
Organopalladium chemistry is a branch of organometallic chemistry that deals with organic palladium compounds and their reactions. Palladium is often used as a catalyst in the reduction of alkenes and alkynes with hydrogen. This process involves the formation of a palladium-carbon covalent bond. Palladium is also prominent in carbon-carbon coupling reactions, as demonstrated in tandem reactions.
The Hiyama coupling is a palladium-catalyzed cross-coupling reaction of organosilanes with organic halides used in organic chemistry to form carbon–carbon bonds. This reaction was discovered in 1988 by Tamejiro Hiyama and Yasuo Hatanaka as a method to form carbon-carbon bonds synthetically with chemo- and regioselectivity. The Hiyama coupling has been applied to the synthesis of various natural products.
The Corey–House synthesis is an organic reaction that involves the reaction of a lithium diorganylcuprate with an organic halide or pseudohalide to form a new alkane, as well as an ill-defined organocopper species and lithium (pseudo)halide as byproducts.
The Negishi coupling is a widely employed transition metal catalyzed cross-coupling reaction. The reaction couples organic halides or triflates with organozinc compounds, forming carbon-carbon bonds (C-C) in the process. A palladium (0) species is generally utilized as the metal catalyst, though nickel is sometimes used. A variety of nickel catalysts in either Ni0 or NiII oxidation state can be employed in Negishi cross couplings such as Ni(PPh3)4, Ni(acac)2, Ni(COD)2 etc.
Organozinc chemistry is the study of the physical properties, synthesis, and reactions of organozinc compounds, which are organometallic compounds that contain carbon (C) to zinc (Zn) chemical bonds.
Transmetalation (alt. spelling: transmetallation) is a type of organometallic reaction that involves the transfer of ligands from one metal to another. It has the general form:
Organocopper chemistry is the study of the physical properties, reactions, and synthesis of organocopper compounds, which are organometallic compounds containing a carbon to copper chemical bond. They are reagents in organic chemistry.
In organic chemistry, the Buchwald–Hartwig amination is a chemical reaction for the synthesis of carbon–nitrogen bonds via the palladium-catalyzed coupling reactions of amines with aryl halides. Although Pd-catalyzed C-N couplings were reported as early as 1983, Stephen L. Buchwald and John F. Hartwig have been credited, whose publications between 1994 and the late 2000s established the scope of the transformation. The reaction's synthetic utility stems primarily from the shortcomings of typical methods for the synthesis of aromatic C−N bonds, with most methods suffering from limited substrate scope and functional group tolerance. The development of the Buchwald–Hartwig reaction allowed for the facile synthesis of aryl amines, replacing to an extent harsher methods while significantly expanding the repertoire of possible C−N bond formation.
A carbometalation is any reaction where a carbon-metal bond reacts with a carbon-carbon π-bond to produce a new carbon-carbon σ-bond and a carbon-metal σ-bond. The resulting carbon-metal bond can undergo further carbometallation reactions or it can be reacted with a variety of electrophiles including halogenating reagents, carbonyls, oxygen, and inorganic salts to produce different organometallic reagents. Carbometalations can be performed on alkynes and alkenes to form products with high geometric purity or enantioselectivity, respectively. Some metals prefer to give the anti-addition product with high selectivity and some yield the syn-addition product. The outcome of syn and anti- addition products is determined by the mechanism of the carbometalation.
In organic chemistry, a cross-coupling reaction is a reaction where two different fragments are joined. Cross-couplings are a subset of the more general coupling reactions. Often cross-coupling reactions require metal catalysts. One important reaction type is this:
Decarboxylative cross coupling reactions are chemical reactions in which a carboxylic acid is reacted with an organic halide to form a new carbon-carbon bond, concomitant with loss of CO2. Aryl and alkyl halides participate. Metal catalyst, base, and oxidant are required.
Tamejiro Hiyama is a Japanese organic chemist. He is best known for his work in developing the Nozaki-Hiyama-Kishi reaction and the Hiyama coupling. He is currently a professor at the Chuo University Research and Development Initiative, and a Professor Emeritus of Kyoto University.
Dialkylbiaryl phosphine ligands are phosphine ligands that are used in homogeneous catalysis. They have proved useful in Buchwald-Hartwig amination and etherification reactions as well as Negishi cross-coupling, Suzuki-Miyaura cross-coupling, and related reactions. In addition to these Pd-based processes, their use has also been extended to transformations catalyzed by nickel, gold, silver, copper, rhodium, and ruthenium, among other transition metals.
Cross electrophile coupling is a type of cross-coupling reaction that occurs between two electrophiles often catalyzed by transition metal catalyst(s). Unlike conventional cross-coupling reactions of an electrophile with an organometallic reagent, the coupling partners in cross electrophile coupling reactions are both electrophiles. Generally, additional reductant to regenerate active catalyst is needed in this reaction.
Miyaura borylation, also known as the Miyaura borylation reaction, is a named reaction in organic chemistry that allows for the generation of boronates from vinyl or aryl halides with the cross-coupling of bis(pinacolato)diboron in basic conditions with a catalyst such as PdCl2(dppf). The resulting borylated products can be used as coupling partners for the Suzuki reaction.
Norio Miyaura was a Japanese organic chemist. He was a professor of graduate chemical engineering at Hokkaido University. His major accomplishments surrounded his work in cross-coupling reactions / conjugate addition reactions of organoboronic acids and addition / coupling reactions of diborons and boranes. He is also the co-author of Cross-Coupling Reactions: A Practical Guide with M. Nomura E. S.. Miyaura was a world-known and accomplished researcher by the time he retired and so, in 2007, he won the Japan Chemical Society Award.
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.




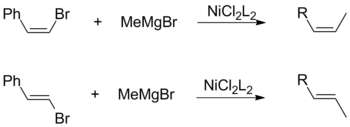

![Chiral ligands for enantioselective Kumada couplings. A: [Methoxyalkyl(ferrocenyl)] monophosphine B: bis-oxazoline Chiral Ligand1.png](http://upload.wikimedia.org/wikipedia/commons/thumb/2/21/Chiral_Ligand1.png/300px-Chiral_Ligand1.png)





