Reported in 1993 by Nicos Petasis as a practical method towards the synthesis of a geometrically pure antifungal agent, naftifine.[1][2][3] In the Petasis reaction, the vinyl group of the organoboronic acid serves as the nucleophile. In comparison to other methods of generating allyl amines, the Petasis reaction tolerates a multifunctional scaffold, with a variety of amines and organoboronic acids as potential starting materials. Additionally, the reaction does not require anhydrous or inert conditions. As a mild, selective synthesis, the Petasis reaction is useful in generating α-amino acids, and is utilized in combinatorial chemistry and drug discovery.[4][5][6][7]
Reaction scope and synthetic applications
The amine is condensed with the carbonyl followed by addition of the boronic acid .[1]
Alpha amino acid synthesis
One of the most attractive features of the Petasis reaction is the stability of the vinyl boronic acids. With the advent of the Suzuki coupling, many are commercially available.
organoboronic acid synthesis
Other methods of generating boronic acids were also reported.[8][9]
A wide variety of functional groups including alcohols, carboxylic acids, and amines are tolerated in the Petasis Reaction. Known substrates that are compatible with reaction conditions include vinylboronate esters, arylboronate esters, and potassium organotrifluoroborates.[10][11][12] Additionally, a variety of substituted amines can be used other than secondary amines. Tertiary aromatic amines, hydrazines, hydroxylamines, sulfonamides, and indoles have all been reported.[13][14][15][16]
Synthesis of allyl amines
Vinyl boronic acids react with the adducts of secondary amines and paraformaldehyde to give tertiary allylamines. The geometry of the double bond of the starting vinyl boronic acid is retained in the final product:[1]
β,γ-unsaturated, N-substituted amino acids are prepared through the condensation of organoboronic acids, boronates, or boronic esters with amines and glyoxylic acids. The highly polar protic solvents Hexafluoroisopropanol (HFIP) can shorten reaction time and improve yield.[17]
PBM coupling to synthesize amino acid with HFIP solvent
Apart from vinyl boronic acids, aryl boronic acids and other heterocyclic derivatives can also be used in Petasis multicomponent coupling. Possible substrate scope includes thienyl, pyridyl, furyl, and benzofuranyl, 1-naphthyl, and aryl groups with either electron-donating or electron-withdrawing substituent.[10]
PBM coupling to synthesize aryl glycine
Racemic Clopidogrel, an antiplatelet agent, was synthesized in two steps using Petasis reaction.[18]
synthesis of clopidogrel via PBM coupling
The Petasis reaction exhibits high degrees of stereocontrol when a chiral amine or aldehyde is used as a substrate. When certain chiral amines, such as (S)-2-phenylglycinol, are mixed with an α-keto acid and vinyl boronic acid at room temperature, the corresponding allylamine is formed as a single diastereomer. Furthermore, enantiomeric purity can be achieved by hydrogenation of the diastereoselective product.
stereoselective alpha amino acids
Unconventional synthesis of carboxylic acids
Apart from amino-acids, PBM reaction can also be used to prepare carboxylic acids, albeit with unconventional mechanisms. In the case of N-substituted indoles as amine equivalent, the reaction begins with the nucleophilic attack of the 3-position of the "N"-substituted indole to electrophilic aldehyde, followed by formation of "ate complex" 1 via the reaction of boronic acid with the carboxylic acid. The intermediate then undergoes dehydration, followed by migration of boronate-alkyl group to furnish the final carboxylic acid product. A wide range of aryl boronic acids is tolerated, while the usage of vinyl boronic acids is not reported. "N"-unsubstituted indoles react very sluggishly under normal reaction conditions, thus confirming the mechanism below.[16]
PBM coupling with N-substituted indole
Tertiary aromatic amines can be used in the Petasis reaction as another equivalent of amine nucleophile. The mechanism is similar to the N-substituted indole case. The reaction is carried out under harsh conditions (24-hr reflux in 1,4-dioxane), but the resultant carboxylic acid is obtained in reasonable yield. Usage of α-ketoacids instead of glyoxylic acid does not diminish yields. 1,3,5-trioxygenated benzene derivatives can also be used in lieu of tertiary aromatic amines.[15]
PBM coupling with trisubstituted aromatic amine
Synthesis of iminodicarboxylic acid derivatives
When used as nitrogen nucleophiles, amino acids can furnish various iminodicarboxylic acid derivatives. High diastereoselectivity is usually observed, and the newly formed stereocenter usually share the same configuration with the starting amino acid. This reaction works well in highly polar solvents (ex. water, ethanol, etc.). Peptides with unprotected nitrogen terminal can also be used as a nitrogen nucleophile equivalent. Petasis and coworkers prepared Enalaprilat, an ACE inhibitor, with this method.[19]
synthesis of Enalaprilat via PBM coupling
Synthesis of peptidomimetic heterocycles
When diamines are used in PBM reactions, heterocycles of various structures, such as piperazinones, benzopiperazinones, and benzodiazepinones, are efficiently prepared. Lactamization reactions are commonly employed to form the heterocycles, usually under strongly acidic conditions.[19]
preparation of Piperazinones, benzopiperazinones, and benzodiazepinones via PBM coupling
Synthesis of amino alcohols
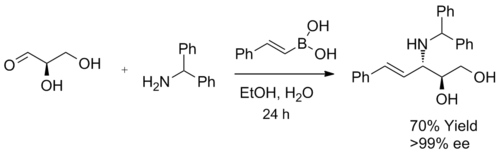
When a α-hydroxy aldehyde is used as a substrate in the synthesis of β-amino alcohols, a single diastereomer is generated. This reaction forms exclusively anti-product, confirmed by 1H NMR spectroscopy. The product does not undergo racemization, and when enantiomerically pure α-hydroxy aldehydes are used, enantiomeric excess can be achieved. It is believed that the boronic acid first reacted with the chiral hydroxyl group, furnishing a nucleophilic alkenyl boronate, followed by face selective, intramolecular migration of the alkenyl group into the electrophilic iminium carbon, forming the desired C–C bond irreversibly.[3]
Stereoselective B amino alcohols
Diastereoselectivity may arise from the reaction of the more stable (and, in this case, more reactive) conformation of the ate complex, where 1,3 allylic strain is minimized.[20][21][22]
diastereoselectivity of amino synthesis_mechanism and transition state
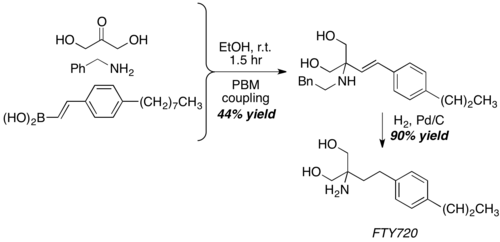
With dihydroxyacetone, a somewhat unconventional aldehyde equivalent, Petasis reaction give the core structure of FTY720, a potent immunosuppressive agent.[23]
synthesis of FTY720
Synthesis of amino polyols and amino sugars
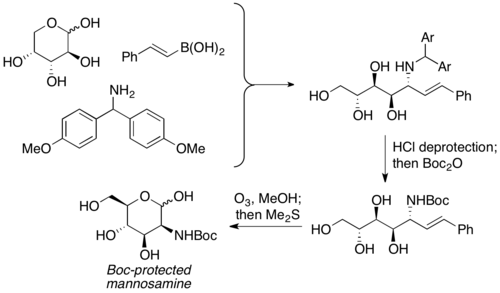
Petasis and coworkers reported the usage of unprotected carbohydrates as the carbonyl component in PBM reactions. It is used as the equivalent of α-hydroxyl aldehydes with pre-existing chirality, and the aminopolyol product is usually furnished with moderate to good yield, with excellent selectivity. A wide variety of carbohydrates, as well as nitrogen nucleophiles (ex. amino acids), can be used to furnish highly stereochemically enriched products. The aminopolyol products can then undergo further reactions to prepare aminosugars. Petasis used this reaction to prepare Boc-protected mannosamine from D-arabinose.[19]
synthesis of Boc-protected mannosammine
Applications in enantioselective synthesis
With chiral amine nucleophile
Generally speaking, when chiral amine is used in Petasis coupling, the stereochemical outcome of Petasis reaction is strongly correlated to the chirality of the amine, and high to excellent diastereoselectivity is observed even without the usage of bulky chiral inducing groups. Chiral benzyl amines,[24] 2-substituted pyrrolidines,[25] and 5-substituted 2-morpholinones[26][27] have been shown to induce good to excellent diastereomeric excess under different Petasis reaction conditions.
diastereoselective PBM coupling with chiral amine
With chiral N-acyliminium ions
Chiral N-acyliminium ion "starting materials" are generally prepared by in-situ dehydration of cyclic hemiaminal. They also carry a chiral hydroxyl group that is in proximity with the iminium carbon; boronic acids react with such chiral hydroxyl groups to form a chiral and electron-rich boronate species, followed by side-selective and intramolecular boronate vinyl/aryl transfer into the iminium carbon. Hence, the reaction is highly diastereoselective, with cis- boronate aryl/vinyl transfer being the predominant pathway. Hydroxypyrrolidines[28] and Hydroxy-γ- and δ-lactams[29] have been shown to react very diastereoselectively, with good to excellent yield. However, such procedures are limited to the usage of vinyl- or electron-rich aryl- boronic acids only.
diastereoselective PBM coupling with chiral N-acyliminium ion
(±)-6-Deoxycastanospermine was prepared in 7 steps from the vinyl boronic ester. The key acyclic precursor to deoxycastanospermine (A) is formed first by condensing vinyl boronic ester 1 with Cbz-protected hydroxy-pyrrolidine 2 with a PBM coupling, followed by dihydroxylation and TBS protection. A then undergo intramolecular cyclization via a one-pot imine formation and reduction sequel, followed by TBS deprotection, to afford (±)-6-deoxycastanospermine.[30]
Deoxycastanospermine synthesis.
With thiourea catalyst
Enantioselective Petasis-type reaction transform quinolines into respective chiral 1,2-dihydroquinolines (product) using alkenyl boronic acids and chiral thiourea catalyst:[31]
Takemoto et al.
Chloroformates are required as electrophilic activating agents. Also, a 1,2-amino alcohol functionality is required on the catalyst for the reaction to proceed stereoselectively.
Takemoto et al. transition state
With chiral biphenols
Chiral α-amino acids with various functionalities are conveniently furnished by mixing alkenyl diethyl boronates, secondary amines, glyoxylates, and chiral biphenol catalyst in toluene in one-pot:[32]
Schaus reaction
This reaction tolerates a wide range of functionalities, both on the sides of alkenyl boronates and the secondary amine: the electron-richness of the substrates does not affect the yield and enantioselectivity, and sterically demanding substrates (dialkylsubstituted alkenyl boronates and amines with α-stereocenter) only compromise enantioselectivity slightly. Reaction rates do vary on a case-by-case basis.[32]
Under the reported condition, boronic acids substrates failed to give any enantioselectivity. Also, 3Å molecular sieve is used in the reaction system. While the authors did not provide the reason for such usage in the paper, it was speculated that 3Å molecular sieves act as water scavenger and prevent the decomposition of alkenyl diethyl boronates into their respective boronic acids. The catalyst could be recycled from the reaction and reused without compromising yield or enantioselectivity.[32]
More recently, Yuan with coworkers from Chengdu Institute of Organic Chemistry, Chinese Academy of Science combined both approaches (chiral thiourea catalyst and chiral biphenol) in a single catalyst, reporting for the first time the catalytic system that is capable of performing enantioselective Petasis reaction between salicylaldehydes, cyclic secondary amines and aryl- or alkenylboronic acids:[33]
Notice that the authors cannot assess syn-1,2-amino alcohol with this method due to intrinsic mechanistic selectivity, and the authors argue that such intrinsic selectivity hampers their ability to access the full matrix of stereoisomeric products for the usage of small molecule screening. In a recent report, Schaus and co-workers reported that syn amino alcohol can be obtained with the following reaction condition, using a chiral dibromo-biphenol catalyst their group developed:[35]
schaus_ACIE_reaction
Although the syn vs. anti diastereomeric ratio ranges from mediocre to good (1.5:1 to 7.5:1), the substrate scope for such reactions remain rather limited, and the diastereoselectivity is found to be dependent on the stereogenic center on the amine starting material.[35]
Petasis reaction and total synthesis
Beau and coworkers assembled the core dihydropyran framework of zanamivir congeners via a combination of PBM reaction and Iron(III)-promoted deprotection-cyclization sequence. A stereochemically defined α-hydroxyaldehyde 2, diallylamine and a dimethylketal-protected boronic acid 1 is coupled to form the acyclic, stereochemically defined amino-alcohol 3, which then undergoes an Iron(III)-promoted cyclization to form a bicyclic dihydropyran 4. Selective opening of the oxazoline portion of the dihydropyran intermediate 4 with water or timethylsilyl azide then furnish downstream products that have structures resembling the Zanamivir family members.[36]
zanamivir core_Beau et al.
Wong and coworkers prepared N-acetylneuraminic acid with a PBM coupling, followed by nitrone-[3+2] cycloaddition. Vinylboronic acid is first coupled with L-arabinose 1 and Bis(4-methoxyphenyl)methanamine 2 to form an stereochemically defined allyl amine 3. Afterwards, the sequence of dipolar cycloaddition, base-mediated N–O bond breakage and hydrolysis then complete the synthesis of N-acetylneuraminic acid.[37]
↑ Petasis, N. A.; Zavialov, I. A. (1997). "A New and Practical Synthesis of -Amino Acids from Alkenyl Boronic Acids". J. Am. Chem. Soc.119 (2): 445–446. doi:10.1021/ja963178n.
↑ Ramadhar, T.R.; Batey, R.A. (2011). "Recent Advances in Nucleophilic Addition Reactions of Organoboronic Acids and Their Derivatives to Unsaturated CN Functionalities". In Hall, D.G. (ed.). Boronic Acids: Preparation and Applications in Organic Synthesis, Medicine and Materials, Second Edition. Wiley-VCH. pp.427–477. doi:10.1002/9783527639328.ch9. ISBN9783527639328.
↑ Hoffmann. R.W.; Dresely, S. (1988), "Preparation of 3-substituted (E)-1-alkenylboronic esters", Synthesis, 1988 (2): 103–106, doi:10.1055/s-1988-27480
↑ Brown, H.C.; Bhat, N.G.; Iyer, R.R. (1991), "A novel route to 1,3-dienyl-2-boronic esters providing simple syntheses of conjugated (E,E)-dienes and conjugated (E)-alkenones", Tetrahedron Lett., 32 (30): 3655–3658, doi:10.1016/s0040-4039(00)79758-9
1 2 Petasis, N.A.; Goodman, A., Zavialov, I.A. (1997), "A new synthesis of α-arylglycines from aryl boronic acids", Tetrahedron, 53 (48): 16463–16470, doi:10.1016/S0040-4020(97)01028-4{{citation}}: CS1 maint: multiple names: authors list (link)
↑ Kabalka, G.W.; Venkataiah, B.; Dong, G. (2004), "The use of potassium alkynyltrifluoroborates in Mannich reactions", Tetrahedron Lett., 45 (4): 729–731, doi:10.1016/j.tetlet.2003.11.049
↑ Tremblay-Morin, J.-P.; Raeppel, S.; Gaudette, F. (2004), "Lewis acid-catalyzed Mannich type reactions with potassium organotrifluoroborate", Tetrahedron Lett., 45 (17): 3471–3474, doi:10.1016/j.tetlet.2004.03.014
↑ Portlock, D.E.; Naskar, D.; West, L.; Li, M. (2002), "Petasis boronic acid-Mannich Reactions of substituted hydrazines: synthesis of α-hydrazino carboxylic acids", Tetrahedron Lett., 43 (38): 6845–6847, doi:10.1016/S0040-4039(02)01511-3
↑ Naskar, D., Roy, A., Seibel, W.L., Portlock, D.E. (2003), "Hydroxylamines and sulfinamide as amine components in the Petasis boronic acid–Mannich reaction: synthesis of N-hydroxy or alkoxy-α-aminocarboxylicacids and N-(tert-butyl sulfinyl)-α-amino carboxylicacids", Tetrahedron Lett., 44 (49): 8865–8868, doi:10.1016/j.tetlet.2003.09.179{{citation}}: CS1 maint: multiple names: authors list (link)
↑ Jourdan, H.; Gouhier, G.; Van Hijfte, L.; Angibaud, P.; Piettre, S. R. (2005). "On the use of boronates in the Petasis reaction". Tetrahedron Lett. 46 (46): 8027–8031. doi:10.1016/j.tetlet.2005.09.060.{{cite journal}}: CS1 maint: multiple names: authors list (link)
↑ Kalinski, C.; Lemoine, H.; Schmidt, J.; Burdack, C.; Kolb, J.; Umkehrer, M.; Ross, G. (2008). "Multicomponent Reactions as a Powerful Tool for Generic Drug Synthesis". Synthesis. 2008 (24): 4007–4011. doi:10.1055/s-0028-1083239.
↑ Davis, A.S.; Pyne, S. G.; Skelton, B. W.; White, A. H. (2004). "Synthesis of putative uniflorine A". J. Org. Chem. 69 (9): 3139–43. doi:10.1021/jo049806y. PMID15104453.
↑ Jiang, B.; Yang, C.-G.; Gu, X.-H. (2001). "A highly stereoselective synthesis of indolyl N-substituted glycines". Tetrahedron Lett. 42 (13): 2545–2547. doi:10.1016/s0040-4039(01)00229-5.
↑ Nanda, K.K.; Trotter, B.W. (2005). "Diastereoselective Petasis Mannich reactions accelerated by hexafluoroisopropanol: a pyrrolidine-derived arylglycine synthesis". Tetrahedron Lett. 46 (12): 2025–8. doi:10.1016/j.tetlet.2005.01.151.
↑ Harwood, L.M.; Currie, G. S.; Drew, M. G. B.; Luke, R. W. A. (1996). "Asymmetry in the boronic acid Mannich reaction: diastereocontrolled addition to chiral iminium species derived from aldehydes and (S)-5-phenylmorpholin-2-one". Chem. Commun. (16): 1953. doi:10.1039/cc9960001953.
↑ Currie, G.S.; Drew, M. G. B.; Harwood, L. M.; Hughes, D. J.; Luke, R. W. A.; Vickers, R. J. (2000). "Chirally templated boronic acid Mannich reaction in the synthesis of optically active α-amino acids". J. Chem. Soc., Perkin Trans. 1 (17): 2982–2990. doi:10.1039/B003067H.
↑ Batey, R.A.; MacKay, D. B.; Santhakumar, V. (1999). "Alkenyl and Aryl BoronatesMild Nucleophiles for the Stereoselective Formation of Functionalized N -Heterocycles". J. Am. Chem. Soc. 121 (21): 5075–5076. Bibcode:1999JAChS.121.5075B. doi:10.1021/ja983801z.
↑ Morgan, I.R.; Yazici, A.; Pyne, S. G. (2008). "Diastereoselective borono-Mannich reactions on cyclic N-acyliminium ions". Tetrahedron. 64 (7): 1409–1419. doi:10.1016/j.tet.2007.11.046.
1 2 Batey, R.A.; MacKay, D.B. (2000). "Total synthesis of (±)-6-deoxycastanospermine: an application of the addition of organoboronates to N-acyliminium ions". Tetrahedron Lett. 41 (51): 9935–9938. doi:10.1016/s0040-4039(00)01790-1.
↑ Yamaoka, Y.; Miyabe, H.; Takemoto, Y. (2007). "Catalytic enantioselective petasis-type reaction of quinolines catalyzed by a newly designed thiourea catalyst". J. Am. Chem. Soc. 129 (21): 6686–7. Bibcode:2007JAChS.129.6686Y. doi:10.1021/ja071470x. PMID17488015.
↑ Naoya Kumagai, Giovanni Muncipinto, Stuart L. Schreiber; Muncipinto; Schreiber (2006). "Short Synthesis of Skeletally and Stereochemically Diverse Small Molecules by Coupling Petasis Condensation Reactions to Cyclization Reactions". Angewandte Chemie International Edition. 45 (22): 3635–3638. doi:10.1002/anie.200600497. PMID16646101.{{cite journal}}: CS1 maint: multiple names: authors list (link)
↑ Soule, J.-F.; Mathieu, A.; Norsikian, S.; Beau, J.-M. (2010). "Coupling the Petasis condensation to an iron(III) chloride-promoted cascade provides a short synthesis of Relenza congeners". Org. Lett. 12 (22): 5322–5325. doi:10.1021/ol102326b. PMID20945892.
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.