
In organic chemistry, a carboxylic acid is an organic acid that contains a carboxyl group attached to an R-group. The general formula of a carboxylic acid is often written as R−COOH or R−CO2H, sometimes as R−C(O)OH with R referring to an organyl group, or hydrogen, or other groups. Carboxylic acids occur widely. Important examples include the amino acids and fatty acids. Deprotonation of a carboxylic acid gives a carboxylate anion.

In organic chemistry, ethers are a class of compounds that contain an ether group—an oxygen atom bonded to two organyl groups. They have the general formula R−O−R′, where R and R′ represent the organyl groups. Ethers can again be classified into two varieties: if the organyl groups are the same on both sides of the oxygen atom, then it is a simple or symmetrical ether, whereas if they are different, the ethers are called mixed or unsymmetrical ethers. A typical example of the first group is the solvent and anaesthetic diethyl ether, commonly referred to simply as "ether". Ethers are common in organic chemistry and even more prevalent in biochemistry, as they are common linkages in carbohydrates and lignin.
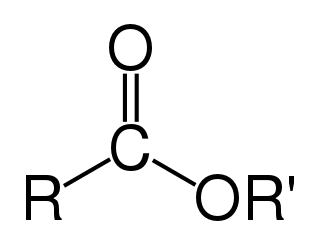
In chemistry, an ester is a functional group derived from an acid in which the hydrogen atom (H) of at least one acidic hydroxyl group of that acid is replaced by an organyl group. Analogues derived from oxygen replaced by other chalcogens belong to the ester category as well. According to some authors, organyl derivatives of acidic hydrogen of other acids are esters as well, but not according to the IUPAC.
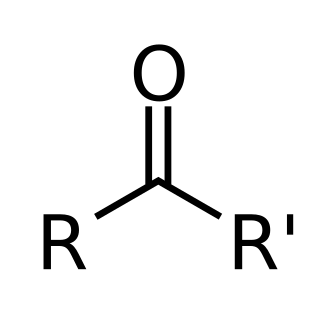
In organic chemistry, a ketone is an organic compound with the structure R−C(=O)−R', where R and R' can be a variety of carbon-containing substituents. Ketones contain a carbonyl group −C(=O)−. The simplest ketone is acetone, with the formula (CH3)2CO. Many ketones are of great importance in biology and in industry. Examples include many sugars (ketoses), many steroids, and the solvent acetone.

In organic chemistry, an aldehyde is an organic compound containing a functional group with the structure R−CH=O. The functional group itself can be referred to as an aldehyde but can also be classified as a formyl group. Aldehydes are a common motif in many chemicals important in technology and biology.

In chemistry, an acyl group is a moiety derived by the removal of one or more hydroxyl groups from an oxoacid, including inorganic acids. It contains a double-bonded oxygen atom and an organyl group or hydrogen in the case of formyl group. In organic chemistry, the acyl group is usually derived from a carboxylic acid, in which case it has the formula R−C(=O)−, where R represents an organyl group or hydrogen. Although the term is almost always applied to organic compounds, acyl groups can in principle be derived from other types of acids such as sulfonic acids and phosphonic acids. In the most common arrangement, acyl groups are attached to a larger molecular fragment, in which case the carbon and oxygen atoms are linked by a double bond.
Hydroboration–oxidation reaction is a two-step hydration reaction that converts an alkene into an alcohol. The process results in the syn addition of a hydrogen and a hydroxyl group where the double bond had been. Hydroboration–oxidation is an anti-Markovnikov reaction, with the hydroxyl group attaching to the less-substituted carbon. The reaction thus provides a more stereospecific and complementary regiochemical alternative to other hydration reactions such as acid-catalyzed addition and the oxymercuration–reduction process. The reaction was first reported by Herbert C. Brown in the late 1950s and it was recognized in his receiving the Nobel Prize in Chemistry in 1979.
A diol is a chemical compound containing two hydroxyl groups. An aliphatic diol may also be called a glycol. This pairing of functional groups is pervasive, and many subcategories have been identified. They are used as protecting groups of carbonyl groups, making them essential in synthesis of organic chemistry.

In organometallic chemistry, organolithium reagents are chemical compounds that contain carbon–lithium (C–Li) bonds. These reagents are important in organic synthesis, and are frequently used to transfer the organic group or the lithium atom to the substrates in synthetic steps, through nucleophilic addition or simple deprotonation. Organolithium reagents are used in industry as an initiator for anionic polymerization, which leads to the production of various elastomers. They have also been applied in asymmetric synthesis in the pharmaceutical industry. Due to the large difference in electronegativity between the carbon atom and the lithium atom, the C−Li bond is highly ionic. Owing to the polar nature of the C−Li bond, organolithium reagents are good nucleophiles and strong bases. For laboratory organic synthesis, many organolithium reagents are commercially available in solution form. These reagents are highly reactive, and are sometimes pyrophoric.
In organic chemistry, dihydroxybenzenes (benzenediols) are organic compounds in which two hydroxyl groups are substituted onto a benzene ring. These aromatic compounds are classed as phenols. There are three structural isomers: 1,2-dihydroxybenzene is commonly known as catechol, 1,3-dihydroxybenzene is commonly known as resorcinol, and 1,4-dihydroxybenzene is commonly known as hydroquinone.
The Cannizzaro reaction, named after its discoverer Stanislao Cannizzaro, is a chemical reaction which involves the base-induced disproportionation of two molecules of a non-enolizable aldehyde to give a primary alcohol and a carboxylic acid.

The Bamford–Stevens reaction is a chemical reaction whereby treatment of tosylhydrazones with strong base gives alkenes. It is named for the British chemist William Randall Bamford and the Scottish chemist Thomas Stevens Stevens (1900–2000). The usage of aprotic solvents gives predominantly Z-alkenes, while protic solvent gives a mixture of E- and Z-alkenes. As an alkene-generating transformation, the Bamford–Stevens reaction has broad utility in synthetic methodology and complex molecule synthesis.
The Baeyer–Villiger oxidation is an organic reaction that forms an ester from a ketone or a lactone from a cyclic ketone, using peroxyacids or peroxides as the oxidant. The reaction is named after Adolf von Baeyer and Victor Villiger who first reported the reaction in 1899.
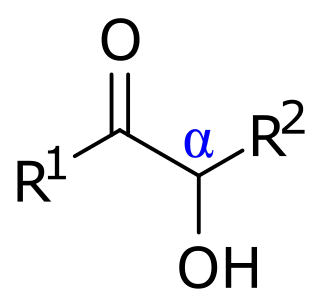
In organic chemistry, acyloins or α-hydroxy ketones are a class of organic compounds of the general form R−C(=O)−CR'(OH)−R", composed of a hydroxy group adjacent to a ketone group. The name acyloin is derived from the fact that they are formally derived from reductive coupling of carboxylic acyl groups. They are one of the two main classes of hydroxy ketones, distinguished by the position of the hydroxy group relative to the ketone; in this form, the hydroxy is on the alpha carbon, explaining the secondary name of α-hydroxy ketone.
The benzilic acid rearrangement is formally the 1,2-rearrangement of 1,2-diketones to form α-hydroxy–carboxylic acids using a base. This reaction receives its name from the reaction of benzil with potassium hydroxide to form benzilic acid. First performed by Justus von Liebig in 1838, it is the first reported example of a rearrangement reaction. It has become a classic reaction in organic synthesis and has been reviewed many times before. It can be viewed as an intramolecular redox reaction, as one carbon center is oxidized while the other is reduced.

The Wolff rearrangement is a reaction in organic chemistry in which an α-diazocarbonyl compound is converted into a ketene by loss of dinitrogen with accompanying 1,2-rearrangement. The Wolff rearrangement yields a ketene as an intermediate product, which can undergo nucleophilic attack with weakly acidic nucleophiles such as water, alcohols, and amines, to generate carboxylic acid derivatives or undergo [2+2] cycloaddition reactions to form four-membered rings. The mechanism of the Wolff rearrangement has been the subject of debate since its first use. No single mechanism sufficiently describes the reaction, and there are often competing concerted and carbene-mediated pathways; for simplicity, only the textbook, concerted mechanism is shown below. The reaction was discovered by Ludwig Wolff in 1902. The Wolff rearrangement has great synthetic utility due to the accessibility of α-diazocarbonyl compounds, variety of reactions from the ketene intermediate, and stereochemical retention of the migrating group. However, the Wolff rearrangement has limitations due to the highly reactive nature of α-diazocarbonyl compounds, which can undergo a variety of competing reactions.

In organic chemistry, vinylogy is the transmission of electronic effects through a conjugated organic bonding system. The concept was introduced in 1926 by Ludwig Claisen to explain the acidic properties of formylacetone and related ketoaldehydes. Formylacetone, technically CH3(C=O)CH2CH=O, only exists in the ionized form CH3(C−O−)=CH−CH=O or CH3(C=O)−CH=CH−O−. Its adjectival form, vinylogous, is used to describe functional groups in which the standard moieties of the group are separated by a carbon–carbon double bond.
The Fleming–Tamao oxidation, or Tamao–Kumada–Fleming oxidation, converts a carbon–silicon bond to a carbon–oxygen bond with a peroxy acid or hydrogen peroxide. Fleming–Tamao oxidation refers to two slightly different conditions developed concurrently in the early 1980s by the Kohei Tamao and Ian Fleming research groups.
The α-ketol rearrangement is the acid-, base-, or heat-induced 1,2-migration of an alkyl or aryl group in an α-hydroxy ketone or aldehyde to give an isomeric product.
Carbonyl oxidation with hypervalent iodine reagents involves the functionalization of the α position of carbonyl compounds through the intermediacy of a hypervalent iodine(III) enolate species. This electrophilic intermediate may be attacked by a variety of nucleophiles or undergo rearrangement or elimination.







