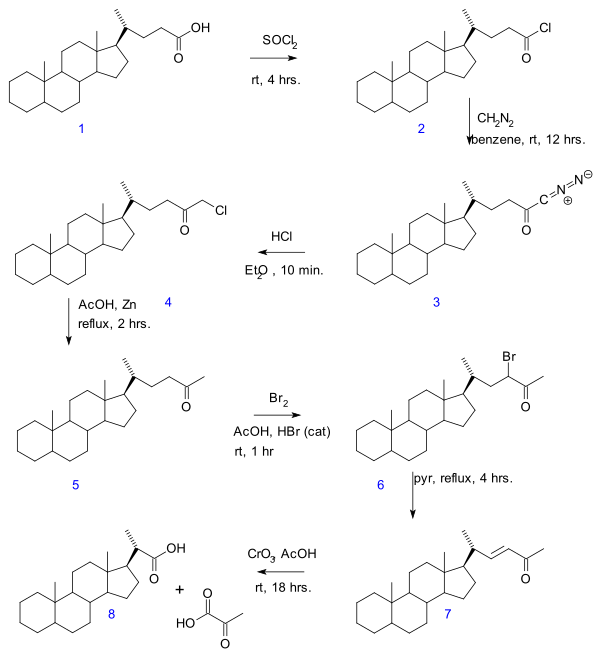
In organic chemistry, a carboxylic acid is an organic acid that contains a carboxyl group attached to an R-group. The general formula of a carboxylic acid is R−COOH or R−CO2H, with R referring to the alkyl, alkenyl, aryl, or other group. Carboxylic acids occur widely. Important examples include the amino acids and fatty acids. Deprotonation of a carboxylic acid gives a carboxylate anion.
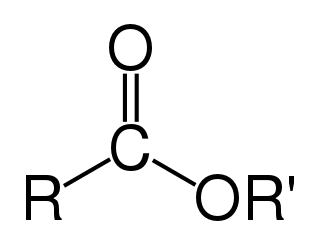
In chemistry, an ester is a compound derived from an oxoacid in which at least one hydroxyl group is replaced by an alkoxy group, as in the substitution reaction of a carboxylic acid and an alcohol. Glycerides are fatty acid esters of glycerol; they are important in biology, being one of the main classes of lipids and comprising the bulk of animal fats and vegetable oils.
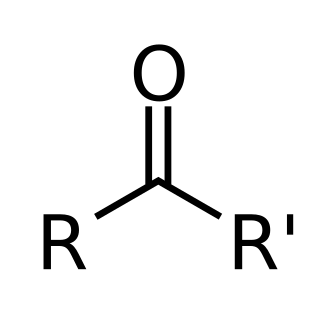
In organic chemistry, a ketone is a functional group with the structure R2C=O, where R can be a variety of carbon-containing substituents. Ketones contain a carbonyl group. The simplest ketone is acetone, with the formula CH3C(O)CH3. Many ketones are of great importance in biology and in industry. Examples include many sugars (ketoses), many steroids, and the solvent acetone.

In organic chemistry, an aldehyde is an organic compound containing a functional group with the structure R−CH=O. The functional group itself can be referred to as an aldehyde but can also be classified as a formyl group. Aldehydes are common and play important roles in the technology and biological spheres.
The Wittig reaction or Wittig olefination is a chemical reaction of an aldehyde or ketone with a triphenyl phosphonium ylide called a Wittig reagent. Wittig reactions are most commonly used to convert aldehydes and ketones to alkenes. Most often, the Wittig reaction is used to introduce a methylene group using methylenetriphenylphosphorane (Ph3P=CH2). Using this reagent, even a sterically hindered ketone such as camphor can be converted to its methylene derivative.

In stereochemistry, a chiral auxiliary is a stereogenic group or unit that is temporarily incorporated into an organic compound in order to control the stereochemical outcome of the synthesis. The chirality present in the auxiliary can bias the stereoselectivity of one or more subsequent reactions. The auxiliary can then be typically recovered for future use.
In organic chemistry, the Arndt–Eistert reaction is the conversion of a carboxylic acid to its homologue. Named for the German chemists Fritz Arndt (1885–1969) and Bernd Eistert (1902–1978), the method entails treating an acid chlorides with diazomethane. It is a popular method of producing β-amino acids from α-amino acids.

The Danishefsky Taxol total synthesis in organic chemistry is an important third Taxol synthesis published by the group of Samuel Danishefsky in 1996 two years after the first two efforts described in the Holton Taxol total synthesis and the Nicolaou Taxol total synthesis. Combined they provide a good insight in the application of organic chemistry in total synthesis.
Glycol cleavage is a specific type of organic chemistry oxidation. The carbon–carbon bond in a vicinal diol (glycol) is cleaved and instead the two oxygen atoms become double-bonded to their respective carbon atoms. Depending on the substitution pattern in the diol, these carbonyls can be either ketones or aldehydes.

The Wolff rearrangement is a reaction in organic chemistry in which an α-diazocarbonyl compound is converted into a ketene by loss of dinitrogen with accompanying 1,2-rearrangement. The Wolff rearrangement yields a ketene as an intermediate product, which can undergo nucleophilic attack with weakly acidic nucleophiles such as water, alcohols, and amines, to generate carboxylic acid derivatives or undergo [2+2] cycloaddition reactions to form four-membered rings. The mechanism of the Wolff rearrangement has been the subject of debate since its first use. No single mechanism sufficiently describes the reaction, and there are often competing concerted and carbene-mediated pathways; for simplicity, only the textbook, concerted mechanism is shown below. The reaction was discovered by Ludwig Wolff in 1902. The Wolff rearrangement has great synthetic utility due to the accessibility of α-diazocarbonyl compounds, variety of reactions from the ketene intermediate, and stereochemical retention of the migrating group. However, the Wolff rearrangement has limitations due to the highly reactive nature of α-diazocarbonyl compounds, which can undergo a variety of competing reactions.
The Étard reaction is a chemical reaction that involves the direct oxidation of an aromatic or heterocyclic bound methyl group to an aldehyde using chromyl chloride. For example, toluene can be oxidized to benzaldehyde.
The total synthesis of quinine, a naturally-occurring antimalarial drug, was developed over a 150-year period. The development of synthetic quinine is considered a milestone in organic chemistry although it has never been produced industrially as a substitute for natural occurring quinine. The subject has also been attended with some controversy: Gilbert Stork published the first stereoselective total synthesis of quinine in 2001, meanwhile shedding doubt on the earlier claim by Robert Burns Woodward and William Doering in 1944, claiming that the final steps required to convert their last synthetic intermediate, quinotoxine, into quinine would not have worked had Woodward and Doering attempted to perform the experiment. A 2001 editorial published in Chemical & Engineering News sided with Stork, but the controversy was eventually laid to rest once and for all when Williams and coworkers successfully repeated Woodward's proposed conversion of quinotoxine to quinine in 2007.

The oxidation of primary alcohols to carboxylic acids is an important oxidation reaction in organic chemistry.
The Lemieux–Johnson or Malaprade–Lemieux–Johnson oxidation is a chemical reaction in which an olefin undergoes oxidative cleavage to form two aldehyde or ketone units. The reaction is named after its inventors, Raymond Urgel Lemieux and William Summer Johnson, who published it in 1956. The reaction proceeds in a two step manner, beginning with dihydroxylation of the alkene by osmium tetroxide, followed by a Malaprade reaction to cleave the diol using periodate. Excess periodate is used to regenerate the osmium tetroxide, allowing it to be used in catalytic amounts. The Lemieux–Johnson reaction ceases at the aldehyde stage of oxidation and therefore produces the same results as ozonolysis.

In the Hooker reaction (1936) an alkyl chain in a certain naphthoquinone is reduced by one methylene unit as carbon dioxide in each potassium permanganate oxidation.
Alcohol oxidation is a class of organic reactions in which the alcohol functional group is converted into another functional group in which carbon carries a higher oxidation state.
An insertion reaction is a chemical reaction where one chemical entity interposes itself into an existing bond of typically a second chemical entity e.g.:

The Jones oxidation is an organic reaction for the oxidation of primary and secondary alcohols to carboxylic acids and ketones, respectively. It is named after its discoverer, Sir Ewart Jones. The reaction was an early method for the oxidation of alcohols. Its use has subsided because milder, more selective reagents have been developed, e.g. Collins reagent.
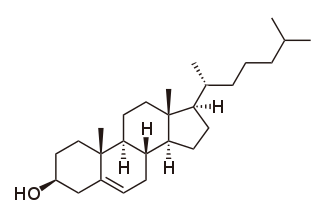
Cholesterol total synthesis in chemistry describes the total synthesis of the complex biomolecule cholesterol and is considered a great scientific achievement. The research group of Robert Robinson with John Cornforth published their synthesis in 1951 and that of Robert Burns Woodward with Franz Sondheimer in 1952. Both groups competed for the first publication since 1950 with Robinson having started in 1932 and Woodward in 1949. According to historian Greg Mulheirn the Robinson effort was hampered by his micromanagement style of leadership and the Woodward effort was greatly facilitated by his good relationships with chemical industry. Around 1949 steroids like cortisone were produced from natural resources but expensive. Chemical companies Merck & Co. and Monsanto saw commercial opportunities for steroid synthesis and not only funded Woodward but also provided him with large quantities of certain chemical intermediates from pilot plants. Hard work also helped the Woodward effort: one of the intermediate compounds was named Christmasterone as it was synthesized on Christmas Day 1950 by Sondheimer.
The Buchner–Curtius–Schlotterbeck reaction is the reaction of aldehydes or ketones with aliphatic diazoalkanes to form homologated ketones. It was first described by Eduard Buchner and Theodor Curtius in 1885 and later by Fritz Schlotterbeck in 1907. Two German chemists also preceded Schlotterbeck in discovery of the reaction, Hans von Pechmann in 1895 and Viktor Meyer in 1905. The reaction has since been extended to the synthesis of β-keto esters from the condensation between aldehydes and diazo esters. The general reaction scheme is as follows: