In chemistry, catalytic resonance theory was developed to describe the kinetics of reaction acceleration using dynamic catalyst surfaces. Catalytic reactions occur on surfaces that undergo variation in surface binding energy and/or entropy, exhibiting overall increase in reaction rate when the surface binding energy frequencies are comparable to the natural frequencies of the surface reaction, adsorption, and desorption.
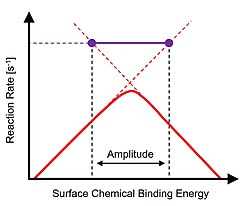
Oscillation of surface binding energy on a Sabatier volcano plot (red) at resonance conditions occurs at the tie line (purple) for maximum average reaction rate
Catalytic resonance theory is constructed on the Sabatier principle of catalysis developed by French chemist Paul Sabatier. In the limit of maximum catalytic performance, the surface of a catalyst is neither too strong nor too weak. Strong binding results in an overall catalytic reaction rate limitation due to product desorption, while weak binding catalysts are limited in the rate of surface chemistry. Optimal catalyst performance is depicted as a 'volcano' peak using a descriptor of the chemical reaction defining different catalytic materials. Experimental evidence of the Sabatier principle was first demonstrated by Balandin in 1960.[1][2]
The concept of catalytic resonance was proposed on dynamic interpretation of the Sabatier volcano reaction plot.[3] As described, extension of either side of the volcano plot above the peak defines the timescales of the two rate-limiting phenomena such as surface reaction(s) or desorption.[4] For binding energy oscillation amplitudes that extend across the volcano peak, the amplitude endpoints intersect the transiently accessible faster timescales of independent reaction phenomena. At the conditions of sufficiently fast binding energy oscillation, the transient binding energy variation frequency matches the natural frequencies of the reaction and the rate of overall reaction achieves turnover frequencies greatly in excess of the volcano plot peak.[5] The single resonance frequency (1/s) of the reaction and catalyst at the selected temperature and oscillation amplitude is identified as the purple tie line; all other applied frequencies are either slower or less efficient.
Theory
The basis of catalytic resonance theory utilizes the transient behavior of adsorption, surface reactions, and desorption as surface binding energy and surface transition states oscillate with time. The binding energy of a single species, i, is described via a temporal functional including square or sinusoidal waves of frequency, fi, and amplitude, dUi:
Response of an A-to-B reversible unimolecular catalytic reaction to a sinusoidal oscillation in surface binding energy.
The two surface species also share the common enthalpy of adsorption, delta δi-j. Specification of the oscillation frequency and amplitude of species i and relating γi-j and δi-j for all other surface species j permits determination of all chemical surface species adsorption enthalpy with time. The transition state energy of a surface reaction between any two species i and j is predicted by the linear scaling relationship of the Bell–Evans–Polanyi principle which relates to the surface reaction enthalpy, ΔHi-j, to the transition state energy, Ea, by parameters α and β with the following relationship:
The oscillating surface and transition state energies of chemical species alter the kinetic rate constants associated with surface reaction, adsorption, and desorption. The surface reaction rate constant of species i converting to surface species j includes the dynamic activation energy:
The resulting surface chemistry kinetics are then described via a surface reaction rate expression containing dynamic kinetic parameters responding to the oscillation in surface binding energy:
,
with k reactions with dynamic activation energy. The desorption rate constant also varies with oscillating surface binding energy by:
.
Implementation of dynamic surface binding energy of a reversible A-to-B reaction on a heterogeneous catalyst in a continuous flow stirred tank reactor operating at 1% conversion of A produces a sinusoidal binding energy in species B as shown.[8] In the transition between surface binding energy amplitude endpoints, the instantaneous reaction rate (i.e., turnover frequency) oscillates over an order of magnitude as a limit cycle solution.
Implications for Chemistry
The effective catalytic rate of a programmable catalyst is a maximum at the applied catalyst resonance frequency
Oscillating binding energies of all surface chemical species introduces periodic instances of transient behavior to the catalytic surface. For slow oscillation frequencies, the transient period is only a small quantity of the oscillation time scale, and the surface reaction achieves a new steady state. However, as the oscillation frequency increases, the surface transient period approaches the timescale of the oscillation, and the catalytic surface remains in a constant transient condition. A plot of the effective catalytic rate of a reaction with respect to applied oscillation frequency identifies the 'resonant' frequency for which the transient conditions of the catalyst surface match the applied frequencies.[9] The catalytic rate at the resonance frequency exists above the Sabatier volcano plot maximum of a static system with average reaction rates as high as five orders of magnitude faster than that achievable by conventional catalysis.
The catalytic reactions of A-to-B and A-to-C can be controlled by applying dynamic binding energy to the surface with varying applied frequency and fixed amplitude starting at varying energies.
Surface binding energy oscillation also occurs to different extent with the various chemical surface species as defined by the γi-j parameter. For any non-unity γi-j system, the asymmetry in the surface energy profile results in conducting work to bias the reaction to a steady state away from equilibrium.[10] Similar to the controlled directionality of molecular machines, the resulting ratchet (device) energy mechanism selectively moves molecules through a catalytic reaction against a free energy gradient.[11]
Application of dynamic binding energy to a surface with multiple catalytic reactions exhibits complex behavior derived from the differences in the natural frequencies of each chemistry; these frequencies are identified by the inverse of the adsorption, desorption, and surface kinetic rate parameters. Considering a system of two parallel elementary reactions of A-to-B and A-to-C that only occur on a surface, the performance of the catalyst under dynamic conditions will result in varying capability for selecting either reaction product (B or C).[12] For the depicted system, both reactions have the same overall thermodynamics and will produce B and C in equal amounts (50% selectivity) at chemical equilibrium. Under normal static catalyst operation, only product B can be produced at selectivities greater than 50% and product C is never favored. However, as shown, the application of surface binding dynamics in the form of a square wave at varying frequency and fixed oscillation amplitude but varying endpoints exhibits the full range of possible reactant selectivity. In the range of 1-10 Hertz, there exists a small island of parameters for which product C is highly selective; this condition is only accessible via dynamics.[13]
Mechanisms of Efficient Dynamic Catalysts
Dynamic catalysts that undergo forced variation of free energy surface during a catalytic reaction are called 'programmable catalysts.' Perturbation of the active site with strain, light, or condensed charge modulates the binding energy of adsorbates and transition states to alter the rate, selectivity, and conversion of a catalytic reaction. New opportunities exist with dynamic catalysts and oscillating free energy surfaces not possible with conventional static active sites. However, these opportunities require energy input to modulate the catalyst, raising the issue of efficiency of a programmable catalyst.[14]
Efficient Programmable Catalyst: A programmable catalyst oscillating between strong and weak binding states efficiently converts purple molecules, A(g), to gold molecules, B(g)
A programmable catalyst oscillating between strong and weak binding energies exhibits positive scaling between reaction intermediates; B* and A* both weaken and strengthen in binding energy simultaneously. Under strong binding conditions, A* readily reacts over the transition state to form B*. For weaking catalyst binding conditions, B* readily desorbs to form B(g), as A(g) immediately adsorbs as A* to restart the catalytic cycle. An efficient programmable catalyst converts molecules from reactants to products with every oscillation of binding energy of the active site, such that most active sites on the catalyst surface produce a product molecule for every catalytic oscillation cycle. Of key importance is the height of the transition state barrier in the weak-binding catalyst state; a high barrier creates a ratchet mechanism, whereby B* is prohibited from reacting backwards to A*.[15]
The efficiency of a programmable catalyst can be determined by the metric of the turnover efficiency (ηTOE). The turnover efficiency compares the difference between the time-averaged dynamic turnover frequency of the reaction (TOFdyn) and the steady state turnover frequency (TOFss) to the applied catalytic oscillation frequency, fapp.
Highly efficient programmable catalysts will exhibit turnover efficiencies close to unity (ηTOE ~ 1), indicating that there is parity between the applied frequency and the catalytic turnover frequency.[16]
Leaky Programmable Catalyst: The programmable catalyst leaky ratchet mechanism occurs when surface molecules are able to preferentially react forwards and backwards relative to desorption.
Of fundamental importance are mechanisms that lead to a reduction in the turnover efficiency. One such catalytic mechanism is the leaky programmable catalyst mechanism. On these dynamic free energy surfaces, the adsorbed surface reactant A* readily reacts to form surface product B* in the strong-binding catalyst state. However, in the weak-binding catalyst state, B* readily reacts backwards to reform A* rather than desorbing to form B(g) in the gas phase. This yields an inefficient programmable catalyst, whereby most input energy to modulate the catalyst between states results in heat generation as molecules interconvert between A* and B*. Programmable catalysts exhibiting the leaky ratchet phenomenon exhibit time-averaged turnover frequencies far from parity with the applied catalytic oscillation frequency, resulting in low turnover efficiency.[17]
Low Surface Participation Programmable Catalyst. A programmable catalytic ratchet exhibits low turnover efficiency due to low participation of the surface in forming surface product, B* (yellow)
An alternative programmable catalytic mechanism leading to reduced turnover efficiency derives from the extent of the surface that participates in the overall reaction. When the programmable catalyst switches to the strong-binding state, A* reacts to B* and equilibrates. However, for systems with comparable free energy of A* and B* in the strong binding state, only a fraction of reactant A* is converted to B*. In this case, only a fraction of surface active sites desorb B* to form B(g) when the catalyst switches to the weak-binding state. Adsorbates of A* that change in binding energy between modulating catalyst states consume energy and release heat without completing the catalytic cycle, yielding an inefficient programmable catalyst.[18]
High turnover efficiency is critical for efficient use of a programmable catalyst. The two key mechanisms leading to lower efficiency, the leaky ratchet and low participating surface mechanisms, can significantly reduce the time-averaged catalytic rate, even orders of magnitude lower than the applied catalyst oscillation frequency.[19]
Characteristics of Dynamic Surface Reactions
Catalytic reactions on surfaces exhibit an energy ratchet that biases the reaction away from equilibrium.[20] In the simplest form, the catalyst oscillates between two states of stronger or weaker binding, which in this example is referred to as 'green' or 'blue,' respectively. For a single elementary reaction on a catalyst oscillating between two states (green & blue), there exists four rate coefficients in total, one forward (k1) and one reverse (k−1) in each catalyst state. The catalyst switches between catalyst states (j of blue or green) with a frequency, f, with the time in each catalyst state, τj, such that the duty cycle, Dj is defined for catalyst state, j, as the fraction of the time the catalyst exists in state j. For the catalyst in the 'blue' state:
The bias of a catalytic ratchet under dynamic conditions can be predicted via a ratchet directionality metric, λ, that can be calculated from the rate coefficients, ki, and the time constants of the oscillation, τi (or the duty cycle).[21] For a catalyst oscillating between two catalyst states (blue and green), the ratchet directionality metric can be calculated:
For directionality metrics greater than 1, the reaction exhibits forward bias to conversion higher than equilibrium. Directionality metrics less than 1 indicate negative reaction bias to conversion less than equilibrium. For more complicated reactions oscillating between multiple catalyst states, j, the ratchet directionality metric can be calculated based on the rate constants and time scales of all states.
The kinetic bias of an independent catalytic ratchet exists for sufficiently high catalyst oscillation frequencies, f, above the ratchet cutoff frequency, fc, calculated as:
For a single independent catalytic elementary step of a reaction on a surface (e.g., A* ↔ B*), the A* surface coverage, θA, can be predicted from the ratchet directionality metric,
Catalytic rate enhancement via dynamic perturbation of surface active sites has been demonstrated experimentally with dynamic electrocatalysis and dynamic photocatalysis. Those results may be explained in the framework of catalytic resonance theory but conclusive evidence is still lacking:
In 1978, the electro-oxidation of formic acid on a platinum electrode was studied under the application of constant potentials and square-wave pulsed potentials. The latter was found to enhance the current density (and thus catalytic activity) by up to 20 times compared to the potentiostatic conditions, with the optimal wave amplitude and frequency of 600 mV and 2000Hz, respectively.[26] In 1988, the oxidation of methanol on a platinum electrode was conducted under pulsed potentials between 0.4 and 1.18 V, resulting in an average current almost 100 times higher than the steady-state current at 0.4 V.[27]
Using the formic acid electro-oxidation reaction, oscillation of the applied electrodynamic potential between 0 and 0.8 volts accelerated the formation rate of carbon dioxide more than an order of magnitude higher (20X) than what was achievable on platinum, the best existing catalyst.[28] The maximum catalytic rate was experimentally observed at a frequency of 100Hz; slower catalytic rates were observed at higher and lower electrodynamic frequencies. The resonant frequency was interpreted as the oscillation between conditions favorable to formic acid decomposition (0 V) and conditions favorable to form CO2 (0.8 V).
The concept of implementing periodic illumination to improve the quantum yield of a typical photocatalytic reaction was first introduced in 1964 by Miller et al. In this work, they showed enhanced photosynthetic efficiency in the conversion of CO2 to O2 when the algal culture was exposed to periodic illumination in a Taylor vortex reactor.[29] Sczechowski et al. later implemented the same approach for heterogeneous photocatalysis in 1993, where they demonstrated 5-fold increment in photoefficiency of formate decomposition by cycling between light and dark conditions with periods of 72 ms and 1.45 s respectively.[30] They hypothesized that upon illumination of the catalyst, there is a critical illumination time during which absorbed photons generate oxidizing species (hvb+) on the surface of the catalyst. The generated species or their intermediates go on to react with substrates on the surface or in the bulk. During dark period, adsorption, desorption, and diffusion generally occurs in the absence of photons. After a critical recovery period in the dark, the photocatalyst can efficiently use photons again when photons are reintroduced. A summary of work involving “dynamic” photocatalysis was provided by Tokode et al. in 2016.[31]
Dynamic promotion of methanol decomposition was demonstrated on 2nm Pt nanoparticles using pulsed light.[32] The rate acceleration to form H2 relative to static illumination was attributed to the selective weakening of adsorbed carbon monoxide, thereby also increasing the quantum efficiency of applied light.
In 2021, Sordello et al.[33] experimentally demonstrated a 50% increase of the quantum yield for the Hydrogen Evolution Reaction (HER) over Pt/TiO2 nanoparticles via formic acid photoreforming under Controlled Period Illumination (CPI).
Implementation of catalyst dynamics has been proposed to occur by additional methods using oscillating light, electric potential, and physical perturbation.[34]
References
↑ Helmut Knözinger; Karl Kochloefl (2005). "Heterogeneous Catalysis and Solid Catalysts". Ullmann's Encyclopedia of Industrial Chemistry. Wiley-VCH Verlag. doi:10.1002/14356007.a05_313. ISBN3527306730.
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.