Elias James Corey is an American organic chemist. In 1990, he won the Nobel Prize in Chemistry "for his development of the theory and methodology of organic synthesis", specifically retrosynthetic analysis. Regarded by many as one of the greatest living chemists, he has developed numerous synthetic reagents, methodologies and total syntheses and has advanced the science of organic synthesis considerably.
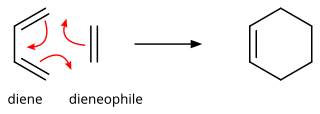
In organic chemistry, the Diels–Alder reaction is a chemical reaction between a conjugated diene and a substituted alkene, commonly termed the dienophile, to form a substituted cyclohexene derivative. It is the prototypical example of a pericyclic reaction with a concerted mechanism. More specifically, it is classified as a thermally-allowed [4+2] cycloaddition with Woodward–Hoffmann symbol [π4s + π2s]. It was first described by Otto Diels and Kurt Alder in 1928. For the discovery of this reaction, they were awarded the Nobel Prize in Chemistry in 1950. Through the simultaneous construction of two new carbon–carbon bonds, the Diels–Alder reaction provides a reliable way to form six-membered rings with good control over the regio- and stereochemical outcomes. Consequently, it has served as a powerful and widely applied tool for the introduction of chemical complexity in the synthesis of natural products and new materials. The underlying concept has also been applied to π-systems involving heteroatoms, such as carbonyls and imines, which furnish the corresponding heterocycles; this variant is known as the hetero-Diels–Alder reaction. The reaction has also been generalized to other ring sizes, although none of these generalizations have matched the formation of six-membered rings in terms of scope or versatility. Because of the negative values of ΔH° and ΔS° for a typical Diels–Alder reaction, the microscopic reverse of a Diels–Alder reaction becomes favorable at high temperatures, although this is of synthetic importance for only a limited range of Diels-Alder adducts, generally with some special structural features; this reverse reaction is known as the retro-Diels–Alder reaction.
A diol is a chemical compound containing two hydroxyl groups. An aliphatic diol is also called a glycol. This pairing of functional groups is pervasive, and many subcategories have been identified.

A trimethylsilyl group (abbreviated TMS) is a functional group in organic chemistry. This group consists of three methyl groups bonded to a silicon atom [−Si(CH3)3], which is in turn bonded to the rest of a molecule. This structural group is characterized by chemical inertness and a large molecular volume, which makes it useful in a number of applications.
A pinacol coupling reaction is an organic reaction in which a carbon–carbon bond is formed between the carbonyl groups of an aldehyde or a ketone in presence of an electron donor in a free radical process. The reaction product is a vicinal diol. The reaction is named after pinacol, which is the product of this reaction when done with acetone as reagent. The reaction is usually a homocoupling but intramolecular cross-coupling reactions are also possible. Pinacol was discovered by Wilhelm Rudolph Fittig in 1859.

The McMurry reaction is an organic reaction in which two ketone or aldehyde groups are coupled to form an alkene using a titanium chloride compound such as titanium(III) chloride and a reducing agent. The reaction is named after its co-discoverer, John E. McMurry. The McMurry reaction originally involved the use of a mixture TiCl3 and LiAlH4, which produces the active reagents. Related species have been developed involving the combination of TiCl3 or TiCl4 with various other reducing agents, including potassium, zinc, and magnesium. This reaction is related to the Pinacol coupling reaction which also proceeds by reductive coupling of carbonyl compounds.

The Danishefsky Taxol total synthesis in organic chemistry is an important third Taxol synthesis published by the group of Samuel Danishefsky in 1996 two years after the first two efforts described in the Holton Taxol total synthesis and the Nicolaou Taxol total synthesis. Combined they provide a good insight in the application of organic chemistry in total synthesis.

The Holton Taxol total synthesis, published by Robert A. Holton and his group at Florida State University in 1994, was the first total synthesis of Taxol.
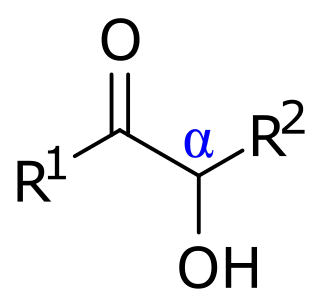
In organic chemistry, acyloins or α-hydroxy ketones are a class of organic compounds of the general form R−C(=O)−CR'(OH)−R", composed of a hydroxy group adjacent to a ketone group. The name acyloin is derived from the fact that they are formally derived from reductive coupling of carboxylic acyl groups. They are one of the two main classes of hydroxy ketones, distinguished by the position of the hydroxy group relative to the ketone; in this form, the hydroxy is on the alpha carbon, explaining the secondary name of α-hydroxy ketone.

The Prins reaction is an organic reaction consisting of an electrophilic addition of an aldehyde or ketone to an alkene or alkyne followed by capture of a nucleophile or elimination of an H+ ion. The outcome of the reaction depends on reaction conditions. With water and a protic acid such as sulfuric acid as the reaction medium and formaldehyde the reaction product is a 1,3-diol (3). When water is absent, the cationic intermediate loses a proton to give an allylic alcohol (4). With an excess of formaldehyde and a low reaction temperature the reaction product is a dioxane (5). When water is replaced by acetic acid the corresponding esters are formed.
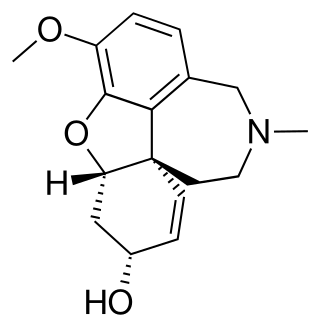
The article concerns the total synthesis of galanthamine, a drug used for the treatment of mild to moderate Alzheimer's disease.

Epothilones are a class of potential cancer drugs. Like taxanes, they prevent cancer cells from dividing by interfering with tubulin, but in early trials, epothilones have better efficacy and milder adverse effects than taxanes.
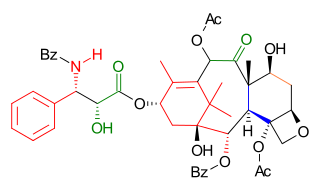
Wender Taxol total synthesis in organic chemistry describes a Taxol total synthesis by the group of Paul Wender at Stanford University published in 1997. This synthesis has much in common with the Holton Taxol total synthesis in that it is a linear synthesis starting from a naturally occurring compound with ring construction in the order A,B,C,D. The Wender effort is shorter by approximately 10 steps.

The Kuwajima Taxol total synthesis by the group of Isao Kuwajima of the Tokyo Institute of Technology is one of several efforts in taxol total synthesis published in the 1990s. The total synthesis of Taxol is considered a landmark in organic synthesis.

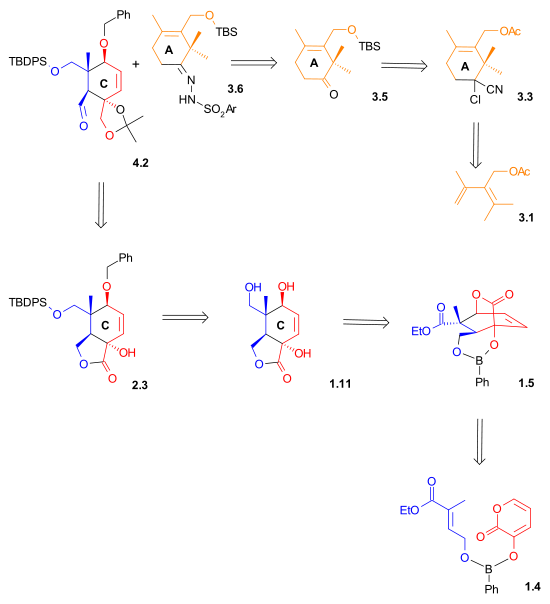
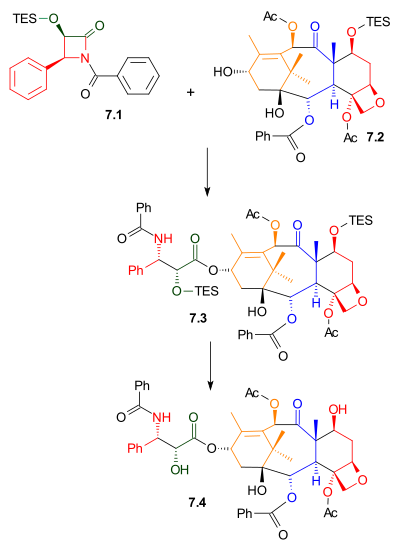
The Mukaiyama taxol total synthesis published by the group of Teruaki Mukaiyama of the Tokyo University of Science between 1997 and 1999 was the 6th successful taxol total synthesis. The total synthesis of Taxol is considered a hallmark in organic synthesis.

Strychnine total synthesis in chemistry describes the total synthesis of the complex biomolecule strychnine. The first reported method by the group of Robert Burns Woodward in 1954 is considered a classic in this research field.

Endiandric acid C, isolated from the tree Endiandra introrsa, is a well characterized chemical compound. Endiadric acid C is reported to have better antibiotic activity than ampicillin.
Alcohol oxidation is a collection of oxidation reactions in organic chemistry that convert alcohols to aldehydes, ketones, carboxylic acids, and esters where the carbon carries a higher oxidation state. The reaction mainly applies to primary and secondary alcohols. Secondary alcohols form ketones, while primary alcohols form aldehydes or carboxylic acids.
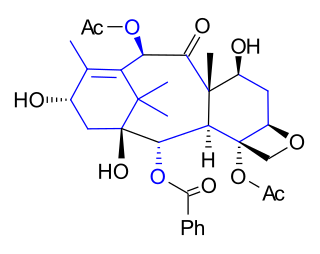
The Takahashi Taxol total synthesis published by Takashi Takahashi in 2006 is one of several methods in taxol total synthesis. The method starts from geraniol and differs from the other 6 published methods that it is a formal synthesis and that it is racemic. A key feature of the published procedure is that several synthetic steps were performed in an automated synthesizer on a scale up to 300 gram and that purification steps were also automated.

Teruaki Mukaiyama was a Japanese organic chemist. One of the most prolific chemists of the 20th century in the field of organic synthesis, Mukaiyama helped establish the field of organic chemistry in Japan after World War II.