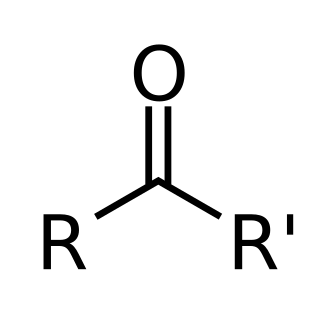
In organic chemistry, a ketone is an organic compound with the structure R−C(=O)−R', where R and R' can be a variety of carbon-containing substituents. Ketones contain a carbonyl group −C(=O)−. The simplest ketone is acetone, with the formula (CH3)2CO. Many ketones are of great importance in biology and in industry. Examples include many sugars (ketoses), many steroids, and the solvent acetone.

In organic chemistry, an aldehyde is an organic compound containing a functional group with the structure R−CH=O. The functional group itself can be referred to as an aldehyde but can also be classified as a formyl group. Aldehydes are a common motif in many chemicals important in technology and biology.

In organic chemistry, an acetal is a functional group with the connectivity R2C(OR')2. Here, the R groups can be organic fragments or hydrogen, while the R' groups must be organic fragments not hydrogen. The two R' groups can be equivalent to each other or not. Acetals are formed from and convertible to aldehydes or ketones and have the same oxidation state at the central carbon, but have substantially different chemical stability and reactivity as compared to the analogous carbonyl compounds. The central carbon atom has four bonds to it, and is therefore saturated and has tetrahedral geometry.

Elias James Corey is an American organic chemist. In 1990, he won the Nobel Prize in Chemistry "for his development of the theory and methodology of organic synthesis", specifically retrosynthetic analysis.
The following outline is provided as an overview of and topical guide to organic chemistry:

A protecting group or protective group is introduced into a molecule by chemical modification of a functional group to obtain chemoselectivity in a subsequent chemical reaction. It plays an important role in multistep organic synthesis.

Dess–Martin periodinane (DMP) is a chemical reagent used in the Dess–Martin oxidation, oxidizing primary alcohols to aldehydes and secondary alcohols to ketones. This periodinane has several advantages over chromium- and DMSO-based oxidants that include milder conditions, shorter reaction times, higher yields, simplified workups, high chemoselectivity, tolerance of sensitive functional groups, and a long shelf life. However, use on an industrial scale is made difficult by its cost and its potentially explosive nature. It is named after the American chemists Daniel Benjamin Dess and James Cullen Martin who developed the reagent in 1983. It is based on IBX, but due to the acetate groups attached to the central iodine atom, DMP is much more reactive than IBX and is much more soluble in organic solvents.

The Nicolaou Taxol total synthesis, published by K. C. Nicolaou and his group in 1994 concerns the total synthesis of taxol. Taxol is an important drug in the treatment of cancer but also expensive because the compound is harvested from a scarce resource, namely the pacific yew.
Dioxolane is a heterocyclic acetal with the chemical formula (CH2)2O2CH2. It is related to tetrahydrofuran (THF) by replacement of the methylene group (CH2) at the 2-position with an oxygen atom. The corresponding saturated 6-membered C4O2 rings are called dioxanes. The isomeric 1,2-dioxolane (wherein the two oxygen centers are adjacent) is a peroxide. 1,3-dioxolane is used as a solvent and as a comonomer in polyacetals.

The Danishefsky Taxol total synthesis in organic chemistry is an important third Taxol synthesis published by the group of Samuel Danishefsky in 1996 two years after the first two efforts described in the Holton Taxol total synthesis and the Nicolaou Taxol total synthesis. Combined they provide a good insight in the application of organic chemistry in total synthesis.

The Holton Taxol total synthesis, published by Robert A. Holton and his group at Florida State University in 1994, was the first total synthesis of Taxol.
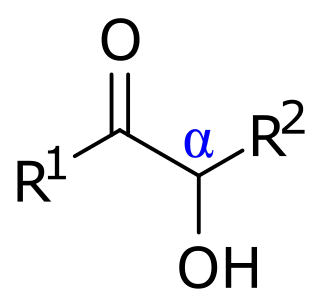
In organic chemistry, acyloins or α-hydroxy ketones are a class of organic compounds of the general form R−C(=O)−CR'(OH)−R", composed of a hydroxy group adjacent to a ketone group. The name acyloin is derived from the fact that they are formally derived from reductive coupling of carboxylic acyl groups. They are one of the two main classes of hydroxy ketones, distinguished by the position of the hydroxy group relative to the ketone; in this form, the hydroxy is on the alpha carbon, explaining the secondary name of α-hydroxy ketone.
The Rubottom oxidation is a useful, high-yielding chemical reaction between silyl enol ethers and peroxyacids to give the corresponding α-hydroxy carbonyl product. The mechanism of the reaction was proposed in its original disclosure by A.G. Brook with further evidence later supplied by George M. Rubottom. After a Prilezhaev-type oxidation of the silyl enol ether with the peroxyacid to form the siloxy oxirane intermediate, acid-catalyzed ring-opening yields an oxocarbenium ion. This intermediate then participates in a 1,4-silyl migration to give an α-siloxy carbonyl derivative that can be readily converted to the α-hydroxy carbonyl compound in the presence of acid, base, or a fluoride source.
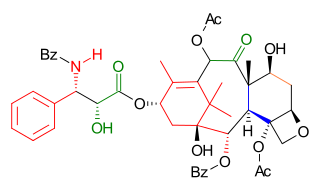
Wender Taxol total synthesis in organic chemistry describes a Taxol total synthesis by the group of Paul Wender at Stanford University published in 1997. This synthesis has much in common with the Holton Taxol total synthesis in that it is a linear synthesis starting from a naturally occurring compound with ring construction in the order A,B,C,D. The Wender effort is shorter by approximately 10 steps.

The Mukaiyama taxol total synthesis published by the group of Teruaki Mukaiyama of the Tokyo University of Science between 1997 and 1999 was the 6th successful taxol total synthesis. The total synthesis of Taxol is considered a hallmark in organic synthesis.

Strychnine total synthesis in chemistry describes the total synthesis of the complex biomolecule strychnine. The first reported method by the group of Robert Burns Woodward in 1954 is considered a classic in this research field.
Alcohol oxidation is a collection of oxidation reactions in organic chemistry that convert alcohols to aldehydes, ketones, carboxylic acids, and esters where the carbon carries a higher oxidation state. The reaction mainly applies to primary and secondary alcohols. Secondary alcohols form ketones, while primary alcohols form aldehydes or carboxylic acids.
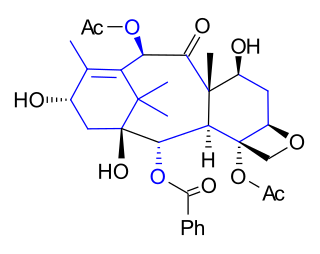
The Takahashi Taxol total synthesis published by Takashi Takahashi in 2006 is one of several methods in taxol total synthesis. The method starts from geraniol and differs from the other 6 published methods that it is a formal synthesis and that it is racemic. A key feature of the published procedure is that several synthetic steps were performed in an automated synthesizer on a scale up to 300 gram and that purification steps were also automated.
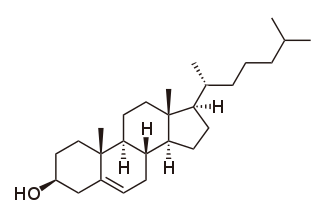
Cholesterol total synthesis in chemistry describes the total synthesis of the complex biomolecule cholesterol and is considered a great scientific achievement. The research group of Robert Robinson with John Cornforth published their synthesis in 1951 and that of Robert Burns Woodward with Franz Sondheimer in 1952. Both groups competed for the first publication since 1950 with Robinson having started in 1932 and Woodward in 1949. According to historian Greg Mulheirn the Robinson effort was hampered by his micromanagement style of leadership and the Woodward effort was greatly facilitated by his good relationships with chemical industry. Around 1949 steroids like cortisone were produced from natural resources but expensive. Chemical companies Merck & Co. and Monsanto saw commercial opportunities for steroid synthesis and not only funded Woodward but also provided him with large quantities of certain chemical intermediates from pilot plants. Hard work also helped the Woodward effort: one of the intermediate compounds was named Christmasterone as it was synthesized on Christmas Day 1950 by Sondheimer.

Sagopilone is a fully synthetic macrolide of the epothilone family with the molecular formula C30H41NO6S. The mechanism of action is similar to taxanes, as they bind to the microtubule and prohibit cell devision. These toxic properties and its possibility to cross the blood-brain barrier makes it a promising cancer medication.





