A protecting group or protective group is introduced into a molecule by chemical modification of a functional group to obtain chemoselectivity in a subsequent chemical reaction. It plays an important role in multistep organic synthesis.
The Wolff–Kishner reduction is a reaction used in organic chemistry to convert carbonyl functionalities into methylene groups. In the context of complex molecule synthesis, it is most frequently employed to remove a carbonyl group after it has served its synthetic purpose of activating an intermediate in a preceding step. As such, there is no obvious retron for this reaction. The reaction was reported by Nikolai Kischner in 1911 and Ludwig Wolff in 1912.

The Corey–Itsuno reduction, also known as the Corey–Bakshi–Shibata (CBS) reduction, is a chemical reaction in which a prochiral ketone is enantioselectively reduced to produce the corresponding chiral, non-racemic alcohol. The oxazaborolidine reagent which mediates the enantioselective reduction of ketones was previously developed by the laboratory of Itsuno and thus this transformation may more properly be called the Itsuno-Corey oxazaborolidine reduction.

The Nicolaou Taxol total synthesis, published by K. C. Nicolaou and his group in 1994 concerns the total synthesis of taxol. Taxol is an important drug in the treatment of cancer but also expensive because the compound is harvested from a scarce resource, namely the pacific yew.

The Wharton olefin synthesis or the Wharton reaction is a chemical reaction that involves the reduction of α,β-epoxy ketones using hydrazine to give allylic alcohols. This reaction, introduced in 1961 by P. S. Wharton, is an extension of the Wolff–Kishner reduction. The general features of this synthesis are: 1) the epoxidation of α,β-unsaturated ketones is achieved usually in basic conditions using hydrogen peroxide solution in high yield; 2) the epoxy ketone is treated with 2–3 equivalents of a hydrazine hydrate in presence of substoichiometric amounts of acetic acid. This reaction occurs rapidly at room temperature with the evolution of nitrogen and the formation of an allylic alcohol. It can be used to synthesize carenol compounds. Wharton's initial procedure has been improved.

The Holton Taxol total synthesis, published by Robert A. Holton and his group at Florida State University in 1994, was the first total synthesis of Taxol.
A Norrish reaction, named after Ronald George Wreyford Norrish, is a photochemical reaction taking place with ketones and aldehydes. Such reactions are subdivided into Norrish type I reactions and Norrish type II reactions. While of limited synthetic utility these reactions are important in the photo-oxidation of polymers such as polyolefins, polyesters, certain polycarbonates and polyketones.
Oppenauer oxidation, named after Rupert Viktor Oppenauer, is a gentle method for selectively oxidizing secondary alcohols to ketones.
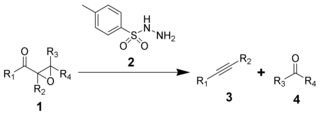
The Eschenmoser fragmentation, first published in 1967, is the chemical reaction of α,β-epoxyketones (1) with aryl sulfonylhydrazines (2) to give alkynes (3) and carbonyl compounds (4). The reaction is named after the Swiss chemist Albert Eschenmoser, who devised it in collaboration with an industrial research group of Günther Ohloff, and applied it to the production of muscone and related macrocyclic musks. The reaction is also sometimes known as the Eschenmoser–Ohloff fragmentation or the Eschenmoser–Tanabe fragmentation as Masato Tanabe independently published an article on the reaction the same year. The general formula of the fragmentation using p-toluenesulfonylhydrazide is:
Wender Taxol total synthesis in organic chemistry describes a Taxol total synthesis by the group of Paul Wender at Stanford University published in 1997. This synthesis has much in common with the Holton Taxol total synthesis in that it is a linear synthesis starting from a naturally occurring compound with ring construction in the order A,B,C,D. The Wender effort is shorter by approximately 10 steps.

The Kuwajima Taxol total synthesis by the group of Isao Kuwajima of the Tokyo Institute of Technology is one of several efforts in taxol total synthesis published in the 1990s. The total synthesis of Taxol is considered a landmark in organic synthesis.

The Mukaiyama taxol total synthesis published by the group of Teruaki Mukaiyama of the Tokyo University of Science between 1997 and 1999 was the 6th successful taxol total synthesis. The total synthesis of Taxol is considered a hallmark in organic synthesis.

An oxocarbeniumion is a chemical species characterized by a central sp2-hybridized carbon, an oxygen substituent, and an overall positive charge that is delocalized between the central carbon and oxygen atoms. An oxocarbenium ion is represented by two limiting resonance structures, one in the form of a carbenium ion with the positive charge on carbon and the other in the form of an oxonium species with the formal charge on oxygen. As a resonance hybrid, the true structure falls between the two. Compared to neutral carbonyl compounds like ketones or esters, the carbenium ion form is a larger contributor to the structure. They are common reactive intermediates in the hydrolysis of glycosidic bonds, and are a commonly used strategy for chemical glycosylation. These ions have since been proposed as reactive intermediates in a wide range of chemical transformations, and have been utilized in the total synthesis of several natural products. In addition, they commonly appear in mechanisms of enzyme-catalyzed biosynthesis and hydrolysis of carbohydrates in nature. Anthocyanins are natural flavylium dyes, which are stabilized oxocarbenium compounds. Anthocyanins are responsible for the colors of a wide variety of common flowers such as pansies and edible plants such as eggplant and blueberry.

Strychnine total synthesis in chemistry describes the total synthesis of the complex biomolecule strychnine. The first reported method by the group of Robert Burns Woodward in 1954 is considered a classic in this research field.
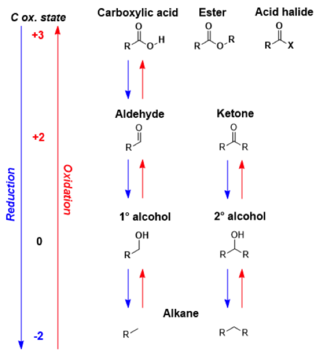
In organic chemistry, carbonyl reduction is the conversion of any carbonyl group, usually to an alcohol. It is a common transformation that is practiced in many ways. Ketones, aldehydes, carboxylic acids, esters, amides, and acid halides - some of the most pervasive functional groups, -comprise carbonyl compounds. Carboxylic acids, esters, and acid halides can be reduced to either aldehydes or a step further to primary alcohols, depending on the strength of the reducing agent. Aldehydes and ketones can be reduced respectively to primary and secondary alcohols. In deoxygenation, the alcohol group can be further reduced and removed altogether by replacement with H.
Reductions with hydrosilanes are methods used for hydrogenation and hydrogenolysis of organic compounds. The approach is a subset of ionic hydrogenation. In this particular method, the substrate is treated with a hydrosilane and auxiliary reagent, often a strong acid, resulting in formal transfer of hydride from silicon to carbon. This style of reduction with hydrosilanes enjoys diverse if specialized applications.

The Narasaka–Prasad reduction, sometimes simply called Narasaka reduction, is a diastereoselective reduction of β-hydroxy ketones to the corresponding syn-dialcohols. The reaction employs a boron chelating agent, such as BBu2OMe, and a reducing agent, commonly sodium borohydride. This protocol was first discovered by Narasaka in 1984.
Borane, also known as borine, is an unstable and highly reactive molecule with the chemical formula BH
3. The preparation of borane carbonyl, BH3(CO), played an important role in exploring the chemistry of boranes, as it indicated the likely existence of the borane molecule. However, the molecular species BH3 is a very strong Lewis acid. Consequently, it is highly reactive and can only be observed directly as a continuously produced, transitory, product in a flow system or from the reaction of laser ablated atomic boron with hydrogen. It normally dimerizes to diborane in the absence of other chemicals.
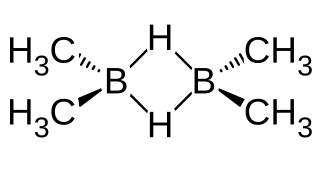
Dimethylborane, (CH3)2BH is the simplest dialkylborane, consisting of a methyl group substituted for a hydrogen in borane. As for other boranes it normally exists in the form of a dimer called tetramethyldiborane or tetramethylbisborane or TMDB ((CH3)2BH)2. Other combinations of methylation occur on diborane, including monomethyldiborane, trimethyldiborane, 1,2-dimethylborane, 1,1-dimethylborane and trimethylborane. At room temperature the substance is at equilibrium between these forms. The methylboranes were first prepared by H. I. Schlesinger and A. O. Walker in the 1930s.

Pinacolborane is the borane with the formula (CH3)4C2O2BH. Often pinacolborane is abbreviated HBpin. It features a boron hydride functional group incorporated in a five-membered C2O2B ring. Like related boron alkoxides, pinacolborane is monomeric. It is a colorless liquid. It features a reactive B-H functional group.