The aldol reaction is a reaction that combines two carbonyl compounds to form a new β-hydroxy carbonyl compound.
A diol is a chemical compound containing two hydroxyl groups. An aliphatic diol is also called a glycol. This pairing of functional groups is pervasive, and many subcategories have been identified.

In organic chemistry, alkenols are a type of reactive structure or intermediate in organic chemistry that is represented as an alkene (olefin) with a hydroxyl group attached to one end of the alkene double bond. The terms enol and alkenol are portmanteaus deriving from "-ene"/"alkene" and the "-ol" suffix indicating the hydroxyl group of alcohols, dropping the terminal "-e" of the first term. Generation of enols often involves deprotonation at the α position to the carbonyl group—i.e., removal of the hydrogen atom there as a proton H+. When this proton is not returned at the end of the stepwise process, the result is an anion termed an enolate. The enolate structures shown are schematic; a more modern representation considers the molecular orbitals that are formed and occupied by electrons in the enolate. Similarly, generation of the enol often is accompanied by "trapping" or masking of the hydroxy group as an ether, such as a silyl enol ether.

A trimethylsilyl group (abbreviated TMS) is a functional group in organic chemistry. This group consists of three methyl groups bonded to a silicon atom [−Si(CH3)3], which is in turn bonded to the rest of a molecule. This structural group is characterized by chemical inertness and a large molecular volume, which makes it useful in a number of applications.

The Nicolaou Taxol total synthesis, published by K. C. Nicolaou and his group in 1994 concerns the total synthesis of taxol. Taxol is an important drug in the treatment of cancer but also expensive because the compound is harvested from a scarce resource, namely the pacific yew.
Silyl ethers are a group of chemical compounds which contain a silicon atom covalently bonded to an alkoxy group. The general structure is R1R2R3Si−O−R4 where R4 is an alkyl group or an aryl group. Silyl ethers are usually used as protecting groups for alcohols in organic synthesis. Since R1R2R3 can be combinations of differing groups which can be varied in order to provide a number of silyl ethers, this group of chemical compounds provides a wide spectrum of selectivity for protecting group chemistry. Common silyl ethers are: trimethylsilyl (TMS), tert-butyldiphenylsilyl (TBDPS), tert-butyldimethylsilyl (TBS/TBDMS) and triisopropylsilyl (TIPS). They are particularly useful because they can be installed and removed very selectively under mild conditions.

The Danishefsky Taxol total synthesis in organic chemistry is an important third Taxol synthesis published by the group of Samuel Danishefsky in 1996 two years after the first two efforts described in the Holton Taxol total synthesis and the Nicolaou Taxol total synthesis. Combined they provide a good insight in the application of organic chemistry in total synthesis.

The Holton Taxol total synthesis, published by Robert A. Holton and his group at Florida State University in 1994, was the first total synthesis of Taxol.
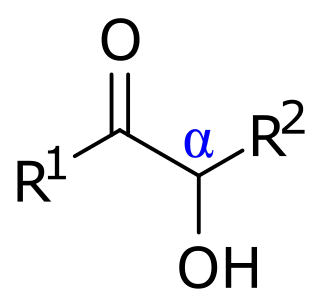
In organic chemistry, acyloins or α-hydroxy ketones are a class of organic compounds of the general form R−C(=O)−CR'(OH)−R", composed of a hydroxy group adjacent to a ketone group. The name acyloin is derived from the fact that they are formally derived from reductive coupling of carboxylic acyl groups. They are one of the two main classes of hydroxy ketones, distinguished by the position of the hydroxy group relative to the ketone; in this form, the hydroxy is on the alpha carbon, explaining the secondary name of α-hydroxy ketone.

Epothilones are a class of potential cancer drugs. Like taxanes, they prevent cancer cells from dividing by interfering with tubulin, but in early trials, epothilones have better efficacy and milder adverse effects than taxanes.
In organosilicon chemistry, silyl enol ethers are a class of organic compounds that share the common functional group R3Si−O−CR=CR2, composed of an enolate bonded to a silane through its oxygen end and an ethene group as its carbon end. They are important intermediates in organic synthesis.
The Rubottom oxidation is a useful, high-yielding chemical reaction between silyl enol ethers and peroxyacids to give the corresponding α-hydroxy carbonyl product. The mechanism of the reaction was proposed in its original disclosure by A.G. Brook with further evidence later supplied by George M. Rubottom. After a Prilezhaev-type oxidation of the silyl enol ether with the peroxyacid to form the siloxy oxirane intermediate, acid-catalyzed ring-opening yields an oxocarbenium ion. This intermediate then participates in a 1,4-silyl migration to give an α-siloxy carbonyl derivative that can be readily converted to the α-hydroxy carbonyl compound in the presence of acid, base, or a fluoride source.

In organic chemistry, the Mukaiyama aldol addition is an organic reaction and a type of aldol reaction between a silyl enol ether and an aldehyde or formate. The reaction was discovered by Teruaki Mukaiyama (1927–2018) in 1973. His choice of reactants allows for a crossed aldol reaction between an aldehyde and a ketone, or a different aldehyde without self-condensation of the aldehyde. For this reason the reaction is used extensively in organic synthesis.
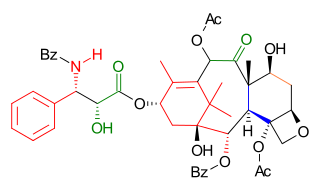
Wender Taxol total synthesis in organic chemistry describes a Taxol total synthesis by the group of Paul Wender at Stanford University published in 1997. This synthesis has much in common with the Holton Taxol total synthesis in that it is a linear synthesis starting from a naturally occurring compound with ring construction in the order A,B,C,D. The Wender effort is shorter by approximately 10 steps.

The Kuwajima Taxol total synthesis by the group of Isao Kuwajima of the Tokyo Institute of Technology is one of several efforts in taxol total synthesis published in the 1990s. The total synthesis of Taxol is considered a landmark in organic synthesis.

Strychnine total synthesis in chemistry describes the total synthesis of the complex biomolecule strychnine. The first reported method by the group of Robert Burns Woodward in 1954 is considered a classic in this research field.

Integrasone is a polyketide natural product, isolated from an unknown fungus, that has been shown to inhibit the HIV-1 integrase enzyme.
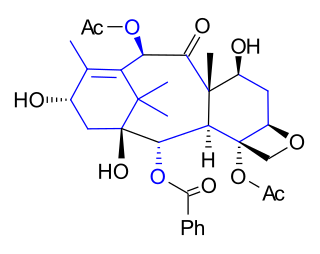
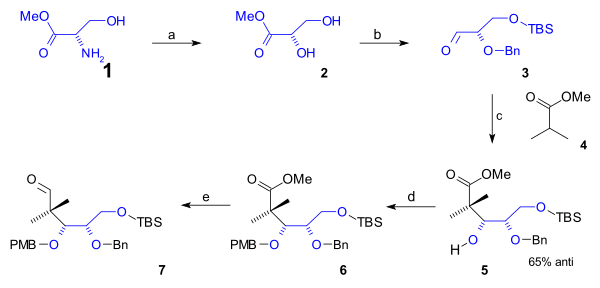
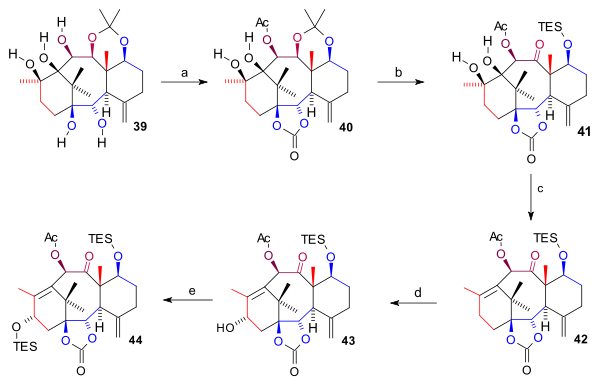
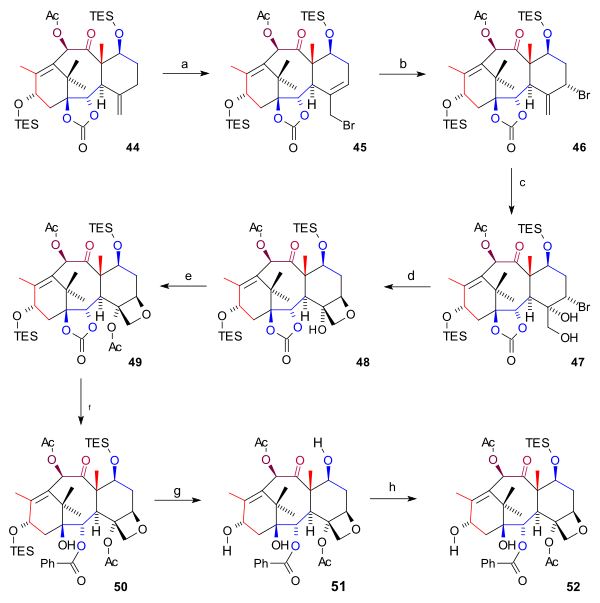
The Takahashi Taxol total synthesis published by Takashi Takahashi in 2006 is one of several methods in taxol total synthesis. The method starts from geraniol and differs from the other 6 published methods that it is a formal synthesis and that it is racemic. A key feature of the published procedure is that several synthetic steps were performed in an automated synthesizer on a scale up to 300 gram and that purification steps were also automated.
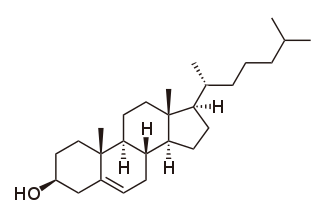
Cholesterol total synthesis in chemistry describes the total synthesis of the complex biomolecule cholesterol and is considered a great scientific achievement. The research group of Robert Robinson with John Cornforth published their synthesis in 1951 and that of Robert Burns Woodward with Franz Sondheimer in 1952. Both groups competed for the first publication since 1950 with Robinson having started in 1932 and Woodward in 1949. According to historian Greg Mulheirn the Robinson effort was hampered by his micromanagement style of leadership and the Woodward effort was greatly facilitated by his good relationships with chemical industry. Around 1949 steroids like cortisone were produced from natural resources but expensive. Chemical companies Merck & Co. and Monsanto saw commercial opportunities for steroid synthesis and not only funded Woodward but also provided him with large quantities of certain chemical intermediates from pilot plants. Hard work also helped the Woodward effort: one of the intermediate compounds was named Christmasterone as it was synthesized on Christmas Day 1950 by Sondheimer.

Teruaki Mukaiyama was a Japanese organic chemist. One of the most prolific chemists of the 20th century in the field of organic synthesis, Mukaiyama helped establish the field of organic chemistry in Japan after World War II.