

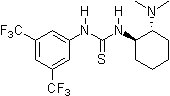
 2003: Takemoto's bifunctional chiral thiourea derivative, catalysis of asymmetric Michael- and Aza-Henry reactions. [13]
2003: Takemoto's bifunctional chiral thiourea derivative, catalysis of asymmetric Michael- and Aza-Henry reactions. [13] 2004: Nagasawa's chiral bis-thiourea organocatalyst, catalysis of asymmetric Baylis-Hillman reactions. [14]
2004: Nagasawa's chiral bis-thiourea organocatalyst, catalysis of asymmetric Baylis-Hillman reactions. [14] 2005: Nagasawa's bifunctional thiourea functionalized guanidine, asymmetric catalysis of Henry(Nitroaldol)reactions. [15]
2005: Nagasawa's bifunctional thiourea functionalized guanidine, asymmetric catalysis of Henry(Nitroaldol)reactions. [15]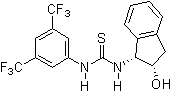 2005: Ricci's chiral thiourea derivative with additional hydroxy-group, enantioselective Friedel-Crafts alkylation of indols with nitroalkenes. [16]
2005: Ricci's chiral thiourea derivative with additional hydroxy-group, enantioselective Friedel-Crafts alkylation of indols with nitroalkenes. [16] 2005: Wei Wang's bifunctional binaphthyl-thiourea derivative, asymmetric catalysis of Morita-Baylis-Hillman reactions. [17]
2005: Wei Wang's bifunctional binaphthyl-thiourea derivative, asymmetric catalysis of Morita-Baylis-Hillman reactions. [17] 2005: Soós's, Connon and Dobson's bifunctional thiourea functionalized Cinchona alkaloid, asymmetric additions of nitroalkanes to chalcones [18] as well as malonates to nitroalkenes [19]
2005: Soós's, Connon and Dobson's bifunctional thiourea functionalized Cinchona alkaloid, asymmetric additions of nitroalkanes to chalcones [18] as well as malonates to nitroalkenes [19] 2006: Yong Tang's chiral bifunctional pyrrolidine-thiourea, enantioselective Michael additions of cyclohexanone to nitroolefins. [20]
2006: Yong Tang's chiral bifunctional pyrrolidine-thiourea, enantioselective Michael additions of cyclohexanone to nitroolefins. [20] 2006: Berkessel's chiral isophoronediamine-derived bisthiourea derivative, catalysis of asymmetric Morita-Baylis-Hillman reactions. [21]
2006: Berkessel's chiral isophoronediamine-derived bisthiourea derivative, catalysis of asymmetric Morita-Baylis-Hillman reactions. [21] 2006: Takemoto's PEG-bound chiral thiourea, asymmetric catalysis of (tandem-) Michael reactions of trans"-β-nitrostyrene, aza-Henry reactions. [22]
2006: Takemoto's PEG-bound chiral thiourea, asymmetric catalysis of (tandem-) Michael reactions of trans"-β-nitrostyrene, aza-Henry reactions. [22] 2007: Kotke/Schreiner, polystyrene-bound, recoverable and reusable thiourea derivative for organocatalytic tetrahydropyranylation of alcohols. [3]
2007: Kotke/Schreiner, polystyrene-bound, recoverable and reusable thiourea derivative for organocatalytic tetrahydropyranylation of alcohols. [3] 2007: Wanka/Schreiner, chiral peptidic adamantane-based thiourea, catalysis of Morita-Baylis-Hillman reactions. [23]
2007: Wanka/Schreiner, chiral peptidic adamantane-based thiourea, catalysis of Morita-Baylis-Hillman reactions. [23] 2007: Takemoto's chelating bifunctional hydroxy-thiourea for enantioselective Petasis-type reaction of quinolines. [24]
2007: Takemoto's chelating bifunctional hydroxy-thiourea for enantioselective Petasis-type reaction of quinolines. [24]
Catalyst-substrate interactions
Hydrogen-bonding between thiourea derivatives and carbonyl substrates involve two hydrogen bonds provided by coplanar amino substituents in the (thio)urea. [2] [3] [4]
[5] Squaramide catalysts engage in double H-bonding interactions and are often superior to thioureas. [6]
Thioureas are often found to be stronger hydrogen-bond donors (i.e., more acidic) than ureas [7] because their amino groups are more positively charged. Quantum chemical analyses revealed that this counterintuitive phenomenon, which is not explainable by the relative electronegativities of O and S, results from the effective steric size of the chalcogen atoms. [8]












