In organic chemistry, a sulfide or thioether is an organosulfur functional group with the connectivity R−S−R' as shown on right. Like many other sulfur-containing compounds, volatile sulfides have foul odors. A sulfide is similar to an ether except that it contains a sulfur atom in place of the oxygen. The grouping of oxygen and sulfur in the periodic table suggests that the chemical properties of ethers and sulfides are somewhat similar, though the extent to which this is true in practice varies depending on the application.
The Friedel–Crafts reactions are a set of reactions developed by Charles Friedel and James Crafts in 1877 to attach substituents to an aromatic ring. Friedel–Crafts reactions are of two main types: alkylation reactions and acylation reactions. Both proceed by electrophilic aromatic substitution.
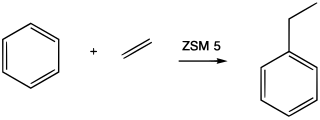
Alkylation is a chemical reaction that entails transfer of an alkyl group. The alkyl group may be transferred as an alkyl carbocation, a free radical, a carbanion, or a carbene. Alkylating agents are reagents for effecting alkylation. Alkyl groups can also be removed in a process known as dealkylation. Alkylating agents are often classified according to their nucleophilic or electrophilic character. In oil refining contexts, alkylation refers to a particular alkylation of isobutane with olefins. For upgrading of petroleum, alkylation produces a premium blending stock for gasoline. In medicine, alkylation of DNA is used in chemotherapy to damage the DNA of cancer cells. Alkylation is accomplished with the class of drugs called alkylating antineoplastic agents.

An enamine is an unsaturated compound derived by the condensation of an aldehyde or ketone with a secondary amine. Enamines are versatile intermediates.

In organometallic chemistry, organolithium reagents are chemical compounds that contain carbon–lithium (C–Li) bonds. These reagents are important in organic synthesis, and are frequently used to transfer the organic group or the lithium atom to the substrates in synthetic steps, through nucleophilic addition or simple deprotonation. Organolithium reagents are used in industry as an initiator for anionic polymerization, which leads to the production of various elastomers. They have also been applied in asymmetric synthesis in the pharmaceutical industry. Due to the large difference in electronegativity between the carbon atom and the lithium atom, the C−Li bond is highly ionic. Owing to the polar nature of the C−Li bond, organolithium reagents are good nucleophiles and strong bases. For laboratory organic synthesis, many organolithium reagents are commercially available in solution form. These reagents are highly reactive, and are sometimes pyrophoric.
Organosulfur chemistry is the study of the properties and synthesis of organosulfur compounds, which are organic compounds that contain sulfur. They are often associated with foul odors, but many of the sweetest compounds known are organosulfur derivatives, e.g., saccharin. Nature is abound with organosulfur compounds—sulfur is vital for life. Of the 20 common amino acids, two are organosulfur compounds, and the antibiotics penicillin and sulfa drugs both contain sulfur. While sulfur-containing antibiotics save many lives, sulfur mustard is a deadly chemical warfare agent. Fossil fuels, coal, petroleum, and natural gas, which are derived from ancient organisms, necessarily contain organosulfur compounds, the removal of which is a major focus of oil refineries.
The Baeyer–Villiger oxidation is an organic reaction that forms an ester from a ketone or a lactone from a cyclic ketone, using peroxyacids or peroxides as the oxidant. The reaction is named after Adolf von Baeyer and Victor Villiger who first reported the reaction in 1899.

Nucleophilic conjugate addition is a type of organic reaction. Ordinary nucleophilic additions or 1,2-nucleophilic additions deal mostly with additions to carbonyl compounds. Simple alkene compounds do not show 1,2 reactivity due to lack of polarity, unless the alkene is activated with special substituents. With α,β-unsaturated carbonyl compounds such as cyclohexenone it can be deduced from resonance structures that the β position is an electrophilic site which can react with a nucleophile. The negative charge in these structures is stored as an alkoxide anion. Such a nucleophilic addition is called a nucleophilic conjugate addition or 1,4-nucleophilic addition. The most important active alkenes are the aforementioned conjugated carbonyls and acrylonitriles.

Isatin, also known as tribulin, is an organic compound derived from indole with formula C8H5NO2. The compound was first obtained by Otto Linné Erdman and Auguste Laurent in 1840 as a product from the oxidation of indigo dye by nitric acid and chromic acids.
The Stollé synthesis is a series of chemical reactions that produce oxindoles from anilines and α-haloacid chlorides.

The Johnson–Corey–Chaykovsky reaction is a chemical reaction used in organic chemistry for the synthesis of epoxides, aziridines, and cyclopropanes. It was discovered in 1961 by A. William Johnson and developed significantly by E. J. Corey and Michael Chaykovsky. The reaction involves addition of a sulfur ylide to a ketone, aldehyde, imine, or enone to produce the corresponding 3-membered ring. The reaction is diastereoselective favoring trans substitution in the product regardless of the initial stereochemistry. The synthesis of epoxides via this method serves as an important retrosynthetic alternative to the traditional epoxidation reactions of olefins.
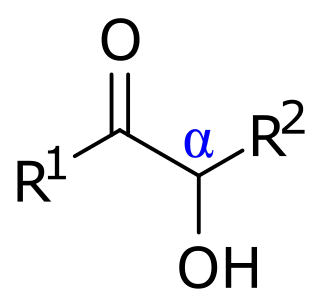
In organic chemistry, acyloins or α-hydroxy ketones are a class of organic compounds of the general form R−C(=O)−CR'(OH)−R", composed of a hydroxy group adjacent to a ketone group. The name acyloin is derived from the fact that they are formally derived from reductive coupling of carboxylic acyl groups. They are one of the two main classes of hydroxy ketones, distinguished by the position of the hydroxy group relative to the ketone; in this form, the hydroxy is on the alpha carbon, explaining the secondary name of α-hydroxy ketone.

The Wolff rearrangement is a reaction in organic chemistry in which an α-diazocarbonyl compound is converted into a ketene by loss of dinitrogen with accompanying 1,2-rearrangement. The Wolff rearrangement yields a ketene as an intermediate product, which can undergo nucleophilic attack with weakly acidic nucleophiles such as water, alcohols, and amines, to generate carboxylic acid derivatives or undergo [2+2] cycloaddition reactions to form four-membered rings. The mechanism of the Wolff rearrangement has been the subject of debate since its first use. No single mechanism sufficiently describes the reaction, and there are often competing concerted and carbene-mediated pathways; for simplicity, only the textbook, concerted mechanism is shown below. The reaction was discovered by Ludwig Wolff in 1902. The Wolff rearrangement has great synthetic utility due to the accessibility of α-diazocarbonyl compounds, variety of reactions from the ketene intermediate, and stereochemical retention of the migrating group. However, the Wolff rearrangement has limitations due to the highly reactive nature of α-diazocarbonyl compounds, which can undergo a variety of competing reactions.

The Stork enamine alkylation involves the addition of an enamine to a Michael acceptor or another electrophilic alkylation reagent to give an alkylated iminium product, which is hydrolyzed by dilute aqueous acid to give the alkylated ketone or aldehyde. Since enamines are generally produced from ketones or aldehydes, this overall process constitutes a selective monoalkylation of a ketone or aldehyde, a process that may be difficult to achieve directly.
In organic chemistry, the Buchwald–Hartwig amination is a chemical reaction for the synthesis of carbon–nitrogen bonds via the palladium-catalyzed coupling reactions of amines with aryl halides. Although Pd-catalyzed C–N couplings were reported as early as 1983, Stephen L. Buchwald and John F. Hartwig have been credited, whose publications between 1994 and the late 2000s established the scope of the transformation. The reaction's synthetic utility stems primarily from the shortcomings of typical methods for the synthesis of aromatic C−N bonds, with most methods suffering from limited substrate scope and functional group tolerance. The development of the Buchwald–Hartwig reaction allowed for the facile synthesis of aryl amines, replacing to an extent harsher methods while significantly expanding the repertoire of possible C−N bond formations.
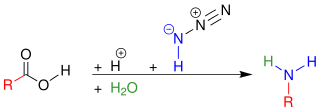
In organic chemistry, the Schmidt reaction is an organic reaction in which an azide reacts with a carbonyl derivative, usually an aldehyde, ketone, or carboxylic acid, under acidic conditions to give an amine or amide, with expulsion of nitrogen. It is named after Karl Friedrich Schmidt (1887–1971), who first reported it in 1924 by successfully converting benzophenone and hydrazoic acid to benzanilide. The intramolecular reaction was not reported until 1991 but has become important in the synthesis of natural products. The reaction is effective with carboxylic acids to give amines (above), and with ketones to give amides (below).
In organic chemistry, the Ei mechanism, also known as a thermal syn elimination or a pericyclic syn elimination, is a special type of elimination reaction in which two vicinal (adjacent) substituents on an alkane framework leave simultaneously via a cyclic transition state to form an alkene in a syn elimination. This type of elimination is unique because it is thermally activated and does not require additional reagents, unlike regular eliminations, which require an acid or base, or would in many cases involve charged intermediates. This reaction mechanism is often found in pyrolysis.

Diisopinocampheylborane is an organoborane that is useful for asymmetric synthesis. This colourless solid is the precursor to a range of related reagents. The compound was reported in 1961 by Zweifel and Brown in a pioneering demonstration of asymmetric synthesis using boranes. The reagent is mainly used for the synthesis of chiral secondary alcohols. The reagent is often depicted as a monomer but like most hydroboranes, it is dimeric with B-H-B bridges.

Indole is an organic compound with the formula C6H4CCNH3. Indoles are derivatives of indole where one or more H's have been replaced by other groups. Indole is classified as an aromatic heterocycle. It has a bicyclic structure, consisting of a six-membered benzene ring fused to a five-membered pyrrole ring. Indoles are widely distributed in nature, most notably as amino acid tryptophan and neurotransmitter serotonin.
Metal-catalyzed C–H borylation reactions are transition metal catalyzed organic reactions that produce an organoboron compound through functionalization of aliphatic and aromatic C–H bonds and are therefore useful reactions for carbon–hydrogen bond activation. Metal-catalyzed C–H borylation reactions utilize transition metals to directly convert a C–H bond into a C–B bond. This route can be advantageous compared to traditional borylation reactions by making use of cheap and abundant hydrocarbon starting material, limiting prefunctionalized organic compounds, reducing toxic byproducts, and streamlining the synthesis of biologically important molecules. Boronic acids, and boronic esters are common boryl groups incorporated into organic molecules through borylation reactions. Boronic acids are trivalent boron-containing organic compounds that possess one alkyl substituent and two hydroxyl groups. Similarly, boronic esters possess one alkyl substituent and two ester groups. Boronic acids and esters are classified depending on the type of carbon group (R) directly bonded to boron, for example alkyl-, alkenyl-, alkynyl-, and aryl-boronic esters. The most common type of starting materials that incorporate boronic esters into organic compounds for transition metal catalyzed borylation reactions have the general formula (RO)2B-B(OR)2. For example, bis(pinacolato)diboron (B2Pin2), and bis(catecholato)diborane (B2Cat2) are common boron sources of this general formula.